2. Part 1: Using the PENCIL CODE
2.1. System requirements
To use the code, you will need the following:
Absolutely needed:
F95 compiler
C compiler
Used heavily (if you don’t have one of these, you will need to adjust many things manually):
a Unix/Linux-type system with make and csh
Perl (remember: if it doesn’t run Perl, it’s not a computer)
The following are dispensable, but enhance functionality in one way or the other:
an MPI implementation (for parallelization on multiprocessor systems)
DX alias OpenDX or data explorer (for 3-D visualization of results)
IDL (for visualization of results; the 7-minute demo license will do for many applications)
2.2. Obtaining the code
The code is now distributed via GitHub repository, where you can either download a tarball, or, preferably, download it via svn or git. In Iran and some other countries, GitHub is not currently available. To alleviate this problem, we have made a recent copy available on http://www.nordita.org/software/pencil-code/. If you want us to update this tarball, please contact us.
To ensure at least some level of stability of the svn/git
versions, a set of test problems (listed in
$PENCIL_HOME/bin/auto-test)
are routinely tested.
This includes all problems in
$PENCIL_HOME/samples.
See S-Testing for details.
2.2.1. Obtaining the code via git or svn
Many machines have svn installed (try
svn -vorwhich svn). On Ubuntu, for example, svn comes under the package namesubversion.The code is now saved under GitHub; git can be obtained in Linux by typing:
sudo apt-get install gitUnless you are a privileged user with write access, you can download the code with the command:
git clone https://github.com/pencil-code/pencil-code.gitor:
svn checkout https://github.com/pencil-code/pencil-code/trunk/ \ pencil-code --username MY_GITHUB_USERNAME
In order to push your changes to the repository, you have to ask the maintainer of the Pencil Code for push access (to become a contributor), or submit a pull request to the maintainer of the code.
Be sure to run
auto-testbefore you check anything back in. It can be very annoying for someone else to figure out what’s wrong, especially if you are just testing something else. At the very least, you should do:pc_auto-test --level=0 --no-pencil-check -CThis allows you to run just the 2 most essential tests, starting with no-modules and then most-modules.
See also Download the Pencil Code for general information on obtaining the code.
2.2.2. Updating via svn or git
Independent of how you installed the code in the first place (from tarball or via svn/git), you can update your version using svn/git. If you have done nontrivial alterations to your version of the code, you ought to be careful about upgrading: although svn/git is an excellent tool for distributed programming, conflicts are quite possible, since many developers may touch many parts of the code while developing it further. Thus, despite the fact that the code is under svn/git, you should probably back up your important changes before upgrading.
Here is the upgrading procedure for git:
Perform a
gitupdate of the tree:unix> git pull \Fix any conflicts you encounter and make sure the examples in the directory
samples/are still working.
Here is the upgrading procedure for svn:
Perform a
svnupdate of the tree:unix> pc_svnup \Fix any conflicts you encounter and make sure the examples in the directory
samples/are still working.
If you have made useful changes, please contact one of the (currently) 10
“Contributors” (listed under GitHub)
who can give you push or check-in permission.
Be sure to have sufficient comments in the code and please follow our
standard coding conventions explained in programming-style.
There is also a script to check and fix the most common style breaks,
pc_codingstyle.
2.2.3. Getting the last validated version
The script pc_svnup accepts arguments -val or -validated, which
means that the current changes on a user’s machine will be merged
into the last working version. This way every user can be sure that
any problems with the code must be due to the current changes done
by this user since the last check-in.
Examples:
$ pc_svnup -src -s -validated
brings all files in src/ under $PENCIL_HOME to the last validated
status, and merges all your changes into this version. This allows you
to work with this, but in order to check in your changes you have to
update everything to the most recent status first, i.e.
$ pc_svnup -src
Your own changes will be merged into this latest version as before.
Note
The functionality of the head of the trunk should be preserved at all times. However, accidents do happen. For the benefit of all
other developers, any errors should be corrected within 1-2 hours.
This is the reason why the code comes with a file
pencil-code/license/developers.txt,
which should contain contact details of all developers.
The pc_svnup -val option allows all other people to stay away
from any trouble.
2.2.4. Getting older versions
You may find that the latest svn version of the code produces errors.
If you have reasons to believe that this is due to changes introduced on
27 November 2008 (to give an example), you can check out the version prior to
this by specifying a revision number with svn update -r #####.
One reason why one cannot always reproduce exactly the same situation too far
back in time is connected with the fact that processor architecture and the
compiler were different, resulting, e.g., in different rounding errors.
2.3. Getting started
To get yourself started, you should run one or several examples which are
provided in one of the samples/ subdirectories.
Note that you will only be able to fully assess the numerical solutions if you
visualize them with IDL, DX, or other tools (see visualization).
2.3.1. Setup
2.3.1.1. Environment settings
The functionality of helper scripts and IDL routines relies on a few
environment variables being set correctly.
The simplest way to achieve this is to go to the top directory of the code
and source one of the two scripts sourceme.csh or sourceme.sh
(depending on the type of shell you are using):
csh> cd pencil-code
csh> source ./sourceme.csh
for tcsh or csh users; or
sh> cd pencil-code
sh> . ./sourceme.sh
for users of bash, Bourne shell, or similar shells. You should get output similar to:
PENCIL_HOME = </home/dobler/f90/pencil-code>
Adding /home/dobler/f90/pencil-code/bin to PATH
Apart from the PATH variable, the environment variable IDL_PATH is set to
something like ./idl:../idl:+$PENCIL_HOME/idl:./data:<IDL_DEFAULT>.
Note
The <IDL_DEFAULT> mechanism does not work for IDL versions 5.2 or
older. In this case, you will have to edit the path manually, or adapt
the sourceme scripts.
Note
If you don’t want to rely on the sourceme scripts’ (quite
heuristic) ability to correctly identify the code’s main directory, you
can set the environment variable PENCIL_HOME explicitly before you
run the source command.
Note
Do not just source the sourceme script from your shell startup
file (~/.cshrc or ~/.bashrc), because it outputs a few
lines of diagnostics for each sub-shell, which will break many applications.
To suppress all output, follow the instructions given in the header
documentation of sourceme.csh and sourceme.sh.
Likewise, output from other files invoked by source should also be suppressed.
Note
The second time you source sourceme, it will not add
anything to your PATH variable.
This is on purpose to avoid cluttering of your environment: you can
source the file as often as you like (in your shell startup script, then
manually and in addition in some script you have written), without
thinking twice.
If, however, at the first sourcing, the setting of PENCIL_HOME was
wrong, this mechanism would keep you from ever adding the right directory
to the PATH.
In this case, you need to first undefine the environment variable
PENCIL_HOME:
csh> unsetenv PENCIL_HOME
csh> source ./sourceme.csh
or
sh> unset PENCIL_HOME
sh> . ./sourceme.sh
Note
If you want to be able to easily handle multiple versions/branches of
Pencil, you can use the modulefile mechanism that is used on most
clusters to load libraries and programs.
Create a file at, say, $HOME/.modulefiles/pencil-local with the
following contents:
#%Module4.6#####################################################################
proc ModulesHelp {} {
global version prefix
puts stderr "\tmodules - loads the modules software"
puts stderr "& application environment"
puts stderr "\n\tThis adds $prefix/* to several of the"
puts stderr "\tenvironment variables."
puts stderr "\n\tVersion $version\n"
}
module-whatis "Environment setup for the Pencil code"
#change the following line according to the location of your local copy of Pencil
setenv PENCIL_HOME $env(HOME)/.software/pencil-code
setenv _sourceme_quiet 1
source-sh bash $env(PENCIL_HOME)/sourceme.sh
unsetenv _sourceme_quiet
To your ~/.bashrc, add:
MODULEPATH=$HOME/.modulefiles:$MODULEPATH
If you now open a new shell and run module avail, you will find the
pencil-local module created above listed as an option.
This requires version 4.6 of the modules program.
2.3.1.2. Linking scripts and source files
With your environment set up correctly, you can now go to the directory
you want to work in and set up subdirectories and links.
This is accomplished by the script pc_setupsrc, which is located in
$PENCIL_HOME/bin and is thus now in your executable path.
For concreteness, let us assume you want to use
samples/conv-slab
as your run directory, i.e., you want to run a three-layer slab model
of solar convection.
You then do the following:
unix> cd samples/conv-slab
unix> pc_setupsrc
src already exists
2 files already exist in src
The script has linked a number of scripts from $PENCIL_HOME/bin,
generated a directory src for the source code and linked the
Fortran source files (plus a few more files) from $PENCIL_HOME/src
to that directory:
unix> ls -F
reference.out src/
start.csh@ run.csh@ getconf.csh@
start.in run.in print.in
2.3.1.3. Adapting Makefile.src
This step requires some input from you, but you only have to do this once for each machine you want to run the code on. See Adapting Makefile.src [obsolete; see Sect.:ref:man1_configuration] for a description of the steps you need to take here.
Note
If you are lucky and use compilers similar to the ones
we have, you may be able to skip this step; but blame yourself if things
don’t compile, then.
If not, you can run make with explicit flags, see
S-make-flags and in particular Table Compiler flags for common compilers.
2.3.1.4. Running make
Next, you run make in the src subdirectory of your run
directory.
Since you are using one of the predefined test problems, the settings in
src/Makefile.local and
src/cparam.local are all reasonable, and you just do:
unix> make
If you have set up the compiler flags correctly, compilation should complete successfully.
2.3.1.5. Choosing a data directory
The code will by default write data like snapshot files to the subdirectory
data of the run directory.
Since this will involve a large volume of IO operations (at least for
large grid sizes), one will normally try to avoid writing the data via
NFS.
The recommended way to set up a data directory is to generate
a corresponding directory on the local disk of the computer you are
running on and (soft-)link it to ./data.
Even if the link is part of an NFS directory, all the IO operations will
be local.
For example, if you have a local disk /scratch, you can do the following:
unix> mkdir -p /scratch/$USER/pencil-data/samples/conv-slab
unix> ln -s /scratch/$USER/pencil-data/samples/conv-slab ./data
This is done automatically by the pc_mkdatadir
command which, in turn, is invoked when making a new run directory with
the pc_newrun command, for example.
Even if you don’t have an NFS-mounted directory (say, on your notebook computer), it is probably still a good idea to have code and data well separated by a scheme like the one described above.
An alternative to symbolic links is to provide a file called
datadir.in in the root of the run directory. This file
should contain one line of text specifying the absolute or relative data
directory path to use. This facility is useful if one wishes to switch
one run directory between different data directories. It is suggested
that in such cases symbolic links are again made in the run directory;
then the datadir.in need contain only a short relative path.
2.3.1.6. Running the code
You are now ready to start the code:
unix> start.csh
Linux cincinnatus 2.4.18-4GB #1 Wed Mar 27 13:57:05 UTC 2002 i686 unknown
Non-MPI version
datadir = data
Fri Aug 8 21:36:43 CEST 2003
src/start.x
CVS: io_dist.f90 v. 1.61 (brandenb ) 2003/08/03 09:26:55
[...]
CVS: start.in v. 1.4 (dobler ) 2002/10/02 20:11:14
nxgrid,nygrid,nzgrid= 32 32 32
thermodynamics: assume cp=1
uu: up-down
piecewise polytropic vertical stratification (lnrho)
init_lnrho: cs2bot,cs2top= 1.450000 0.3333330
e.g., for ionization runs: cs2bot,cs2top not yet set
piecewise polytropic vertical stratification (ss)
start.x has completed successfully
0.070u 0.020s 0:00.14 64.2% 0+0k 0+0io 180pf+0w
Fri Aug 8 21:36:43 CEST 2003
This runs src/start.x to construct an initial condition based on
the parameters set in start.in.
This initial condition is stored in data/proc0/var.dat (and
:data/proc1/var.dat, etc., if you run the multiprocessor version).
It is fair to say that this is now a rather primitive routine; see
pencil-code/idl/read for various reading routines.
You can then visualize the data using standard IDL language.
If you visualize the profiles using IDL (see below),
the result should bear some resemblance to Fig-pvert1, but with
different values in the ghost zones (the correct values are set at
run-time only) and a simpler velocity profile.
Now we run the code:
unix> run.csh
This executes src/run.x and carries out nt time steps,
where nt and other run-time parameters are specified in run.in.
On a decent PC (1.7 GHz), 50 time steps take about 10 seconds.
The relevant part of the code’s output looks like:
--it----t-------dt-------urms----umax----rhom------ssm-----dtc----dtu---dtnu---dtchi-
0 0.34 6.792E-03 0.0060 0.0452 14.4708 -0.4478 0.978 0.013 0.207 0.346
10 0.41 6.787E-03 0.0062 0.0440 14.4707 -0.4480 0.978 0.013 0.207 0.345
20 0.48 6.781E-03 0.0064 0.0429 14.4705 -0.4481 0.977 0.012 0.207 0.345
30 0.54 6.777E-03 0.0067 0.0408 14.4703 -0.4482 0.977 0.012 0.207 0.345
40 0.61 6.776E-03 0.0069 0.0381 14.4702 -0.4482 0.977 0.011 0.207 0.346
The columns list:
it: the number of the current time stept: the timedt: the time stepurms: the rms velocity,urms = sqrt(<u^2>)umax: the maximum velocity,umax = max |u|rhom: the mean density,rhom = <rho>ssm: the mean entropy,ssm = <s>/cpdtc: the time step in units of the acoustic Courant step,dtc = dt * cs0 / dx_mindtu: the time step in units of the advective time step,dtu = dt / (c_delta_t * dx / max|u|)dtnu: the time step in units of the viscous time step,dtnu = dt / (c_delta_t_v * dx^2 / nu_max)dtchi: the time step in units of the conductive time step,dtchi = dt / (c_delta_t_v * dx^2 / chi_max)
The entries in this list can be added, removed or reformatted in the file
print.in (see Sects. Diagnostic output and S-print.in-params).
The output is also saved in data/time_series.dat
and should be identical to the content of reference.out.
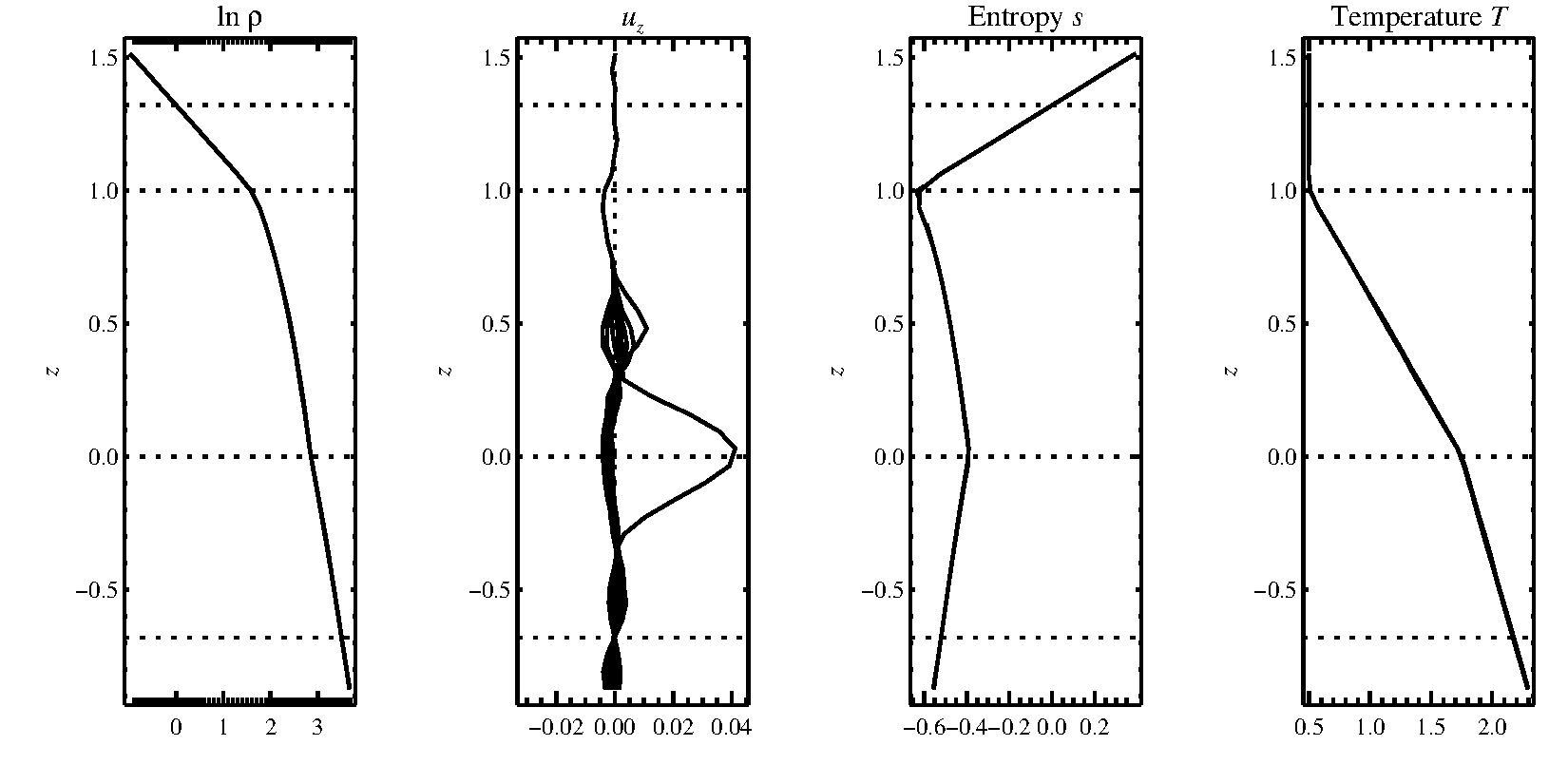
Fig. 2.1 Stratification of the three-layer convection model in
samples/conv-slab after 50 timesteps (t=0.428).
Shown are (from left to right) density rho, vertical velocity u_z,
entropy s/cp and temperature T as functions of the
vertical coordinate z for about ten different vertical lines in the
computational box.
The dashed lines denote domain boundaries:
z < -0.68 is the lower ghost zone (points have no physical significance);
-0.68 < z < 0 is a stably stratified layer (ds/dz > 0);
0 < z < 1 is the unstable layer (ds/dz < 0);
1 < z < 1.32 is the isothermal top layer;
z > 1.32 is the upper ghost zone (points have no physical significance).
If you have IDL, you can visualize the stratification with (see
Sect. IDL for details):
unix > idl
IDL > pc_read_var,obj=var,/trimall
IDL > tvscl,var,uu(*,*,0,0)
which shows u_x in the xy plane through the first
meshpoint in the z direction.
The same can be achieved using Python
(see Sect. Python Runtime Services for details) with:
unix > ipython3 # (or 'ipython', or just 'python')
python > import pencil as pc
python > from matplotlib import pylab as plt
python > var = pc.read.var(trimall=True)
python > plt.imshow(var.uu[0, 0, :, : ].T, origin='lower')
Note
If you want to run the code with MPI, you will probably need to
adapt getconf.csh, which defines the commands and flags used to
run MPI jobs (and which is sourced by the scripts start.csh and
run.csh).
Try:
csh -v getconf.csh
or
csh -x getconf.csh
to see how getconf.csh makes its decisions. You would add a
section for the host name of your machine with the particular settings.
Since getconf.csh is linked from the central directory
pencil-code/bin, your changes will be
useful for all your other runs too.
2.3.2. Further tests
There are a number of other tests in the samples/ directory.
You can use the script bin/auto-test to automatically run
these tests and have the output compared to reference results.
2.4. Code structure
2.4.1. Directory tree
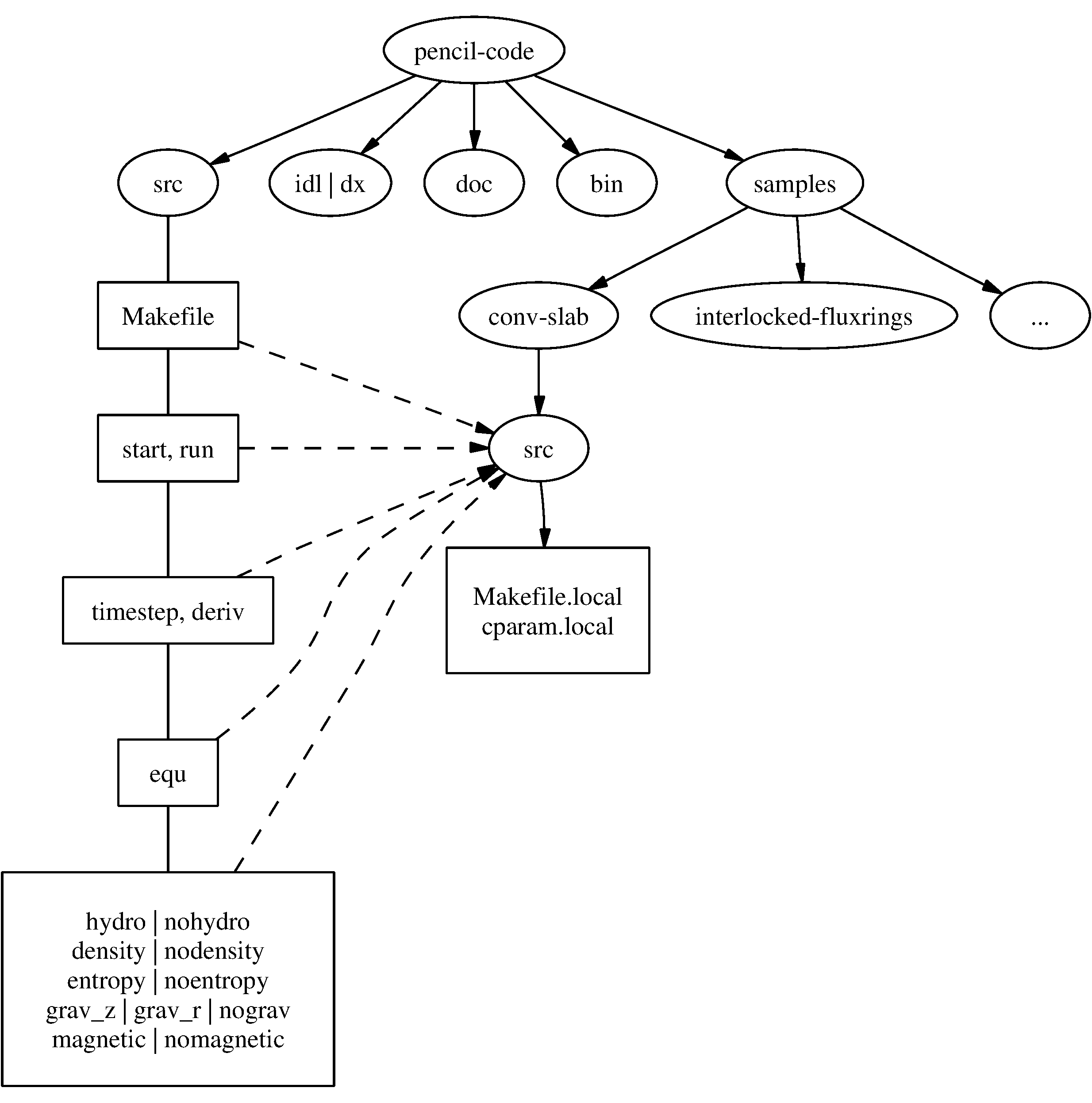
Fig. 2.2 The basic structure of the code
The overall directory structure of the code is shown in Fig. 2.2.
Under pencil-code/, there are currently the following files and directories:
bin/ config/ doc/ idl/ license/ perl/ samples/ sourceme.sh utils/
bugs/ dx/ lib/ misc/ README sourceme.csh src/ www/
Almost all of the source code is contained in the directory src/,
but in order to encapsulate individual applications, the code is compiled
separately for each run in a local directory src/ below the
individual run directory, like
e.,g.~:file:samples/conv-slab/src/.
It may be a good idea to keep your own runs also under SVN or CVS
(which is older than but similar to SVN), but this would normally be a
different repository. On the machine where you are running the code, you
may want to check them out into a subdirectory of pencil-code/.
For example, we have our own runs in a repository called pencil-runs, so we do:
unix> cd $PENCIL_HOME
unix> svn co runs pencil-runs
In this case, runs/ contains individual run directories,
grouped in classes (like spher/ for spherical calculations, or
kinematic/ for kinematic dynamo simulations).
The current list of classes in our own pencil-runs/ repository is
1d-tests/ disc/ kinematic/ rings/
2d-tests/ discont/ Misc/ slab_conv/
3d-tests/ discussion/ OLD/ test/
buoy_tube/ forced/ pass_only/
convstar/ interstellar/ radiation/
The directory forced/ contains some forced turbulence runs (both
magnetic and nonmagnetic);
gravz/ contains runs with vertical gravity;
rings/ contains decaying MHD problems (interlocked flux rings as
initial condition, for example);
and kinematic/ contains kinematic dynamo problems where the
hydrodynamics is turned off entirely.
The file samples/README should contain an up-to-date list and
short description of the individual classes.footnote{Our
pencil-runs/ directory also contains runs that were
done some time ago. Occasionally, we try to update these, especially if we
have changed names or other input conventions.}
The subdirectory src/ of each run directory contains a few local
configuration files (currently these are Makefile.local and
cparam.local) and possibly ctimeavg.local.
To compile the samples, links the files *.f90,
*.c and Makefile.src need to be linked from the top
src/ directory to the local directory ./src.
These links are set up by the script
pc_setupsrc when used in the root of a run directory.
General-purpose visualization routines for IDL or DX are in the
directories idl/ and dx/, respectively.
There are additional and more specialized IDL directories in the
different branches under pencil-runs/.
The directory doc/ contains this manual;
bin/ contains a number of utility scripts (mostly written in
C-shell and Perl), and in particular the start.csh,
run.csh, and getconf.csh scripts.
The .svn/ directory is used (you guessed it) by SVN, and is
not normally directly accessed by the user;
bugs/, finally, is used by us for internal purposes.
The files sourceme.csh and sourceme.sh will set up some
environment variables — in particular PATH — and aliases/shell
functions for your convenience.
If you do not want to source one of these files, you need to make sure
your IDL path is set appropriately (provided you want to use
IDL) and you will need to address the scripts from bin/ with their explicit path name, or adjust your PATH
manually.
2.4.2. Basic concepts
2.4.2.1. Data access in pencils
Unlike the CRAY computers that dominated supercomputing in the 80s and early 90s, all modern computers have a cache that constitutes a significant bottleneck for many codes. This is the case if large three-dimensional arrays are constantly used within each time step, which has the obvious advantage of working on long arrays and allows vectorization of elementary machine operations. This approach also implies conceptual simplicity of the code and allows extensive use of the intuitive F90 array syntax. However, a more cache-efficient way of coding is to calculate an entire time step (or substep of a multi-stage time-stepping scheme) only along a one-dimensional pencil of data within the numerical grid. This technique is more efficient for modern RISC processors: on Linux PCs and SGI workstations, for example, we have found a speed-up by about 60% in some cases. An additional advantage is a drastic reduction in temporary storage for auxiliary variables within each time step.
2.4.2.2. Modularity
Each run directory has a file src/Makefile.local in
which you choose certain modules [1], which tell the code whether or not entropy, magnetic fields,
hydrodynamics, forcing, etc.should be invoked.
For example, the settings for forced turbulent MHD simulations are
HYDRO = hydro
DENSITY = density
ENTROPY = noentropy
MAGNETIC = magnetic
GRAVITY = nogravity
FORCING = forcing
MPICOMM = nompicomm
GLOBAL = noglobal
IO = io_dist
FOURIER = nofourier
This file will be processed by make and the settings are thus
assignments of make variables.
Apart from the physics modules (equation of motion: yes, density
[pressure]: yes, entropy equation: no, magnetic fields: yes, gravity: no,
forcing: yes), a few technical modules can also be used or deactivated; in
the example above, these are MPI (switched off), additional global
variables (none), input/output (distributed), and FFT (not used).
The table in Sect.~:ref:Tab-modules in the Appendix lists all currently available modules.
Note that most of these make variables must be set, but they
will normally obtain reasonable default values in Makefile (so you
only need to set the non-standard ones in Makefile.local).
It is by using this switching mechanism through make that we achieve
high flexibility without resorting to excessive amounts of cryptic
preprocessor directives or other switches within the code.
Many possible combinations of modules have already been tested and examples are part of the distribution, but you may be interested in a combination which was never tried before and which may not work yet, since the modules are not fully orthogonal. In such cases, we depend on user feedback for fixing problems and documenting the changes for others.
2.4.3. Files in the run directories
2.4.3.1. start.in, run.in, print.in
These files specify the startup and runtime parameters (see
Sects. Start and run parameters and S-all-run-params), and the list of
diagnostic variables to print (see Diagnostic output).
They specify the setup of a given simulation and are kept under
svn svn in the individual samples/ directories.
You may want to check for the correctness of these configuration files by
issuing the command pc_configtest.
2.4.3.2. datadir.in
If this file exists, it must contain the name of an existing directory,
which will be used as data directory,
i.e., the directory where all results are written.
If datadir.in does not exist, the data directory is data/.
2.4.3.3. sn_series.in`
Formatted file containing the times and locations at which future supernova
events will occur, using same format as sn_series.dat when lSN_list.
(Only needed by the interstellar module.)
2.4.3.4. reference.out
If present, reference.out contains the output you should obtain in
the given run directory, provided you have not changed any parameters.
To see whether the results of your run are OK, compare time_series.dat to
reference.out:
unix> diff data/time_series.dat reference.out
2.4.3.5. start.csh, run.csh, getconf.csh` [obsolete; see Sect. Configuring the code to compile and run on your computer]
These are links to $PENCIL_HOME/bin.
You will be constantly using the scripts start.csh and
run.csh to initialize the code.
Things that are needed by both (like the name of the mpirun
executable, MPI options, or the number of processors) are located in
getconf.csh, which is never directly invoked.
2.4.3.6. src/
The src/ directory contains two local files,
src/Makefile.local and
src/cparam.local, which allow the user to choose
individual modules (see Modularity) and to set parameters like the
grid size and the number of processors for each direction.
These two files are part of the setup of a given simulation and are kept
under svn in the individual samples/ directories.
The file src/cparam.inc is automatically generated by
the script mkcparam and contains the number of fundamental
variables for a given setup.
All other files in src/ are either links to source files (and
Makefile.src) in the $PENCIL_HOME/src directory,
or object and module files generated by the compiler.
2.4.3.7. data/
This directory (the name of which will actually be overwritten by the first
line of datadir.in, if that file is present; see datadir-in)
contains the output from the code:
2.4.3.7.1. data/dim.dat
The global array dimensions.
2.4.3.7.2. data/legend.dat
The header line specifying the names of the diagnostic variables in
time_series.dat.
2.4.3.7.3. data/time_series.dat
Time series of diagnostic variables (also printed to stdout). You can use this file directly for plotting with Gnuplot, IDL, Xmgrace or similar tools (see also S-Visualization).
2.4.3.7.4. data/tsnap.dat, data/tvid.dat
Time when the next snapshot VAR$N$ or animation slice should be
taken.
2.4.3.7.5. data/params.log
Keeps a log of all your parameters: start.x writes the startup
parameters to this file, run.x appends the runtime parameters and
appends them anew, each time you use the RELOAD mechanism (see RELOAD, STOP and SAVE files).
2.4.3.7.6. data/param.nml
Complete set of startup parameters, printed as Fortran namelist.
This file is read in by run.x (this is how values of startup
parameters are propagated to run.x) and by IDL (if you use it).
2.4.3.7.7. data/param2.nml
Complete set of runtime parameters, printed as Fortran namelist. This file is read by IDL (if you use it).
2.4.3.7.8. data/index.pro
Can be used as include file in IDL and contains the column in which
certain variables appear in the diagnostics file (time_series.dat).
It also contains the positions of variables in the VAR$N$ files.
These positions depend on whether entropy or noentropy, etc,
are invoked.
This is a temporary solution and the file may disappear in future
releases.
2.4.3.7.9. data/sn_series.dat
Time series of SN explosions locations and diagnostics. Can be plotted
using same machinery as for time_series.dat and stored as
sn_series.in to replicate series in subsequent experiments.
(Only needed by the interstellar module.)
2.4.3.7.10. [proc$N]data/proc0, data/proc1, …
These are the directories containing data from the individual processors.
So after running an MPI job on two processors, you will have the
two directories proc0 and proc1.
Each of the directories can contain the following files:
var.dat- binary file containing the latest snapshot;VAR$N$- binary file containing individual snapshot number $N$;dim.dat- ASCII file containing the array dimensions as seen by the given processor;time.dat- ASCII file containing the time corresponding tovar.dat(not actually used by the code, unless you use theio_mpiodist.f90module);grid.dat- binary file containing the part of the grid seen by the given processor;seed.dat- the random seed for the next time step (saved for reasons of reproducibility). For multi-processor runs with velocity forcing, the filesproc$N$/seed.datmust all contain the same numbers, because globally coherent waves of given wavenumber are used;X.xy,X.xz,X.yz- two-dimensional sections of variable X, where X stands for the corresponding variable. The current list includes:bx.xy bx.xz by.xy by.xz bz.xy bz.xz divu.xy lnrho.xz ss.xz ux.xy ux.xz uz.xy uz.xz
Each processor writes its own slice, so these need t be reassembled if one wants to plot a full slice.
2.5. Using the code
2.5.1. Configuring the code to compile and run on your computer
Note
We recommend to use the procedure described here, rather than the old method described in Sects. Adapting Makefile.src [obsolete; see Sect.:ref:man1_configuration] and start.csh, run.csh, getconf.csh` [obsolete; see Sect. man1_configuration].
2.5.1.1. Quick instructions
You may compile with a default compiler-specific configuration:
Single-processor using the GNU compiler collection:
unix> pc_build -f GNU-GCCMulti-processor using GNU with MPI support:
unix> pc_build -f GNU-GCC_MPI
Many compilers are supported already; please refer to the available config
files in $PENCIL_HOME/config/compilers/*.conf, e.g.,
Intel.conf and Intel_MPI.conf.
If you have to set up some compiler options specific to a certain host system you work on,
or if you like to create a host-specific configuration file so that you can
simply execute pc_build without any options,
you can clone an existing host-file, just include an existing
compiler configuration, and simply only add the options you need.
A good example of a host-file is
$PENCIL_HOME/config/hosts/IWF/host-andromeda-GNU_Linux-Linux.conf.
You may save a clone under $PENCIL_HOME/config/hosts/<ID>.conf,
where <ID> is to be replaced by the output of pc_build -i.
This will be the new default for pc_build.
Another way to specify the default is setting the environment variable PENCIL_CONFIG_FILES
appropriately.
If you don’t know what this was all about, read on.
In essence, configuration, compiling and running the code work like this:
Create a configuration file for your computer’s host ID.
Compile the code using
pc_build.Run the code using
pc_run.
In the following, we will discuss the essentials of this scheme.
Exhaustive documentation is available with the commands
perldoc Pencil::ConfigFinder and perldoc PENCIL::ConfigParser.
2.5.1.2. Locating the configuration file
Commands like pc_build and pc_run use the Perl module
Pencil::ConfigFinder to locate an appropriate configuration file
and Pencil::ConfigParser to read and interpret it.
When you use ConfigFinder on a given computer, it constructs a
host ID for the system it is running on, and looks for a file
host_ID.conf` in any subdirectory of $PENCIL_HOME/config/hosts.
For example, if the host ID is workhorse.pencil.org, ConfigFinder would
consider the file
$PENCIL_HOME/config/hosts/pencil.org/workhorse.pencil.org.conf.
Note
The location in the tree under hosts/ is irrelevant, which allows
you to group related hosts by institution, owner, hardware, etc.
Note
ConfigFinder actually uses the following search path:
./config$PENCIL_HOME/config-local$HOME/.pencil/config$PENCIL_HOME/config
This allows you to override part of the config/ tree globally on
the given file system, or locally for a particular run directory, or for
one given copy of the Pencil Code.
This search path is used both for locating the configuration file for
your host ID and for locating included files (see below).
The host ID is constructed based on information that is easily available for your system. The algorithm is as follows:
Most commands using
ConfigFinderhave a--host-id(sometimes abbreviated as-H) option to explicitly set the host ID.If the environment variable
PENCIL_HOST_IDis set, its value is used.If any of the files
./host-ID$PENCIL_HOME/host-ID$HOME/.pencil/host-ID
exists, its first line is used.
If
ConfigFindercan get hold of a fully qualified host name, that is used as host ID.Else, a combination of host name, operating system name and possibly some other information characterizing the system is used.
If no config file for the host ID is found, the current operating system name is tried as fallback host ID.
To see which host IDs are tried (up to the first one for which a configuration file is found), run:
unix> pc_build --debug-config
This command will tell you the host-ID of the system that you are using:
unix> pc_build -i
2.5.1.3. Structure of configuration files
It is strongly recommended to include in a user’s configuration file one of the preset compiler suite configuration files. Then, only minor options need to be set by a user, e.g., the optimization flags. One of those user configuration files looks rather simple:
# Simple config file. Most files don't need more.
%include compilers/GNU-GCC
or if you prefer a different compiler:
# Simple Intel compiler suite config file.
%include compilers/Intel
A more complex file (using MPI with additional options) would look like this:
# More complex config file.
%include compilers/GNU-GCC_MPI
%section Makefile
MAKE_VAR1 = -j4 # joined compilation with four threads
FFLAGS += -O3 -Wall -fbacktrace # don't redefine, but append with '+='
%endsection Makefile
%section runtime
mpiexec = mpirun # some MPI backends need a redefinition of mpiexec
%endsection runtime
%section environment
SCRATCH_DIR=/var/tmp/$USER
%endsection environment
Adding “_MPI” to a compiler suite’s name is usually sufficient to use MPI.
Note
We strongly advise not to mix Fortran- and C-compilers from different manufacturers or versions by manually including multiple separate compiler configurations.
Note
We strongly advise to use at maximum the optimization levels ‘-O2’ for the Intel compiler and ‘-O3’ for all other compilers. Higher optimization levels implicate an inadequate loss of precision.
The .conf files consist of the following elements:
Comments: A
#sign and any text following it on the same line are ignored.Sections: There are three sections:
Makefile for setting
makeparametersruntime for adding compiler flags used by
pc_runenvironment shell environment variables for compilation and running
Include statements: An
%include ...statement is recursively replaced by the contents of the files it points to. [2]The included path gets a
.confsuffix appended. Included paths are typically “absolute”, e.g.,:%include os/Unix
will include the file
os/Unix.confin the search path listed above (typically from$PENCIL_HOME/config). However, if the included path starts with a dot, it is a relative path, so:%include ./Unix
will only search in the directory where the including file is located.
Assignments: Statements like
FFLAGS += -O3ormpiexec=mpirunare assignments and will set parameters that are used bypc_build/makeorpc_run.Lines ending with a backslash
\are continuation lines.If possible, one should always use incremental assignments, indicated by using a
+=sign instead of=, instead of redefining certain flags.Thus:
CFLAGS += -O3 CFLAGS += -I../include -Wall
is the same as:
CFLAGS = $(CFLAGS) -O3 -I../include -Wall
2.5.1.4. Compiling the code
Use the pc_build command to compile the code:
unix> pc_build # use default compiler suite
unix> pc_build -f Intel_MPI # specify a compiler suite
unix> pc_build -f os/GNU_Linux,mpi/open-mpi # explicitly specify config files
unix> pc_build -l # use same config files as in last call of pc_build
unix> pc_build VAR=something # set variables for the makefile
unix> pc_build --cleanall # remove generated files
The third example circumvents the whole host ID mechanism by explicitly
instructing pc_build which configuration files to use.
In the fourth example, pc_build will apply the same configuration files as in its last invocation.
They are stored in src/.config-files, which is automatically written, but can also be manually modified.
The fifth example shows how to define extra variables (VAR=something) for the execution of the Makefile.
See pc_build --help for a complete list of options.
2.5.1.5. Running the code
Use the pc_run command to run the code:
unix> pc_run # start if necessary, then run
unix> pc_run start
unix> pc_run run
unix> pc_run start run^3 # start, then run 3 times
unix> pc_run start run run run # start, then run 3 times
unix> pc_run ^3 # start if necessary, then run 3 times
See pc_run --help for a complete list of options.
2.5.1.6. Testing the code
The pc_auto-test command uses pc_build for compiling and
pc_run for running the tests.
Here are a few useful options:
unix> pc_auto-test
unix> pc_auto-test --no-pencil-check # suppress pencil consistency check
unix> pc_auto-test --max-level=1 # run only tests in level 0 and 1
unix> pc_auto-test --time-limit=2m # kill each test after 2 minutes
See pc_auto-test --help for a complete list of options.
The pencil-test script will use pc_auto-test if given the
--use-pc_auto-test or -b option:
unix> pencil-test --use-pc_auto-test
unix> pencil-test -b # ditto
unix> pencil-test -b \
-Wa,--max-level=1,--no-pencil-check # quick pencil test
See pencil-test --help for a complete list of options, and section S-Testing for more details.
2.5.2. Adapting Makefile.src [obsolete; see Sect.:ref:man1_configuration]
By default, one should use the above described configuration mechanism for compilation. If for whatever reason one needs to work with a modified Makefile, there is a mechanism for picking the right set of compiler flags based on the hostname.
To give you an idea of how to add your own machines, let us assume you have
several Linux boxes running a compiler f95 that needs the options
-O2 -u, while one of them, Janus, runs a compiler f90
which needs the flags -O3 and requires the additional
options -lmpi -llam for MPI.
The Makefile.src you need will have the following section:
### Begin machine dependent
## IRIX64:
[...] (leave as it is in the original Makefile)
## OSF1:
[...] (leave as it is in the original Makefile)
## Linux:
[...] (leave everything from original Makefile and add:)
#FC=f95
#FFLAGS=-O2 -u
#FC=f90 #(Janus)
#FFLAGS=-O3 #(Janus)
#LDMPI=-lmpi -llam #(Janus)
## SunOS:
[...] (leave as it is in the original Makefile)
## UNICOS/mk:
[...] (leave as it is in the original Makefile)
## HI-UX/MPP:
[...] (leave as it is in the original Makefile)
## AIX:
[...] (leave as it is in the original Makefile)
### End machine dependent
Note
There is a script for adapting the Makefile: adapt-mkfile.
In the example above, #(Janus) is not a comment, but marks
this line to be activated (uncommented) by adapt-mkfile if your
hostname (uname -n) matches Janus or janus (capitalization is irrelevant).
You can combine machine names with a vertical bar: a line containing
#(onsager|Janus) will be activated on both Janus and Onsager.
2.5.2.1. Experimenting with compiler flags
If you want to experiment with compiler flags, or if you
want to get things running without setting up the machine-dependent
section of the Makefile, you can set make variables at the
command line in the usual manner:
src> make FC=f90 FFLAGS='-fast -u'
This will use the compiler f90 and the flags -fast -u for both compilation and linking.
Table Table 2.1 summarizes flags we use for common compilers.
Compiler |
FC |
FFLAGS |
CC |
CFLAGS |
|---|---|---|---|---|
Unix/POSIX: |
||||
GNU |
gfortran |
-O3 |
gcc |
-O3 -DFUNDERSC=1 |
Intel |
ifort |
-O2 |
icc |
-O3 -DFUNDERSC=1 |
PGI |
pgf95 |
-O3 |
pgcc |
-O3 -DFUNDERSC=1 |
G95 |
g95 |
-O3 -fno-second-underscore |
gcc |
-O3 -DFUNDERSC=1 |
Absoft |
f95 |
-O3 -N15 |
gcc |
-O3 -DFUNDERSC=1 |
IBM XL |
xlf95 |
-qsuffix=f=f90 -O3 |
xlc |
-O3 -DFUNDERSC=1 |
outdated: |
||||
IRIX Mips |
f90 |
-64 -O3 -mips4 |
cc |
-O3 -64 -DFUNDERSC=1 |
Compaq |
f90 |
-fast -O3 |
cc |
-O3 -DFUNDERSC=1 |
2.5.2.1.1. Changing the resolution
It is advisable to produce a new run directory each time you run a new case.
(This does not include restarts from an old run, of course.)
If you have a 32^3 run in some directory runA_32a, then go to
its parent directory, i.e.:
runA_32a> cd ..
forced> pc_newrun runA_32a runA_64a
forced> cd runA_64a/src
forced> vi cparam.local
and edit the cparam.local for the new resolution.
If you have ever wondered why we don’t do dynamic allocation of the main variable (f) array, the main reason it that with static allocation the compiler can check whether we are out of bounds.
2.5.2.2. Using a non-equidistant grid
We introduce a non-equidistant grid \(z_i\) (by now, this is also implemented for the other directions) as a function \(z(\zeta)\) of an equidistant grid \(\zeta_i\) with grid spacing \(\Delta \zeta = 1\).
The way the parameters are handled, the box size and position are
not changed when you switch to a non-equidistant grid, i.e.,
they are still determined by xyz0 and Lxyz.
The first and second derivatives can be calculated by:
which can be written somewhat more compactly using the inverse function \(\zeta(z)\):
Internally, the code uses the quantities:
and stores them in data/proc$N$/grid.dat.
The parameters lequidist (a 3-element logical array), grid_func,
coeff_grid (a ≥ 2-element real array) are used to choose a
non-equidistant grid and define the function \(z(\zeta)\).
So far, one can choose between grid_function='sinh',
grid_function='linear' (equidistant grid for testing), and
grid_function='step-linear'.
2.5.2.2.1. The sinh profile:
For grid_function='sinh', the function \(z(\zeta)\) is given by:
where \(z_0\) and \(z_0+L_z\) are the lowest and uppermost levels, \(\zeta_1, \zeta_2\) are the \(\zeta\) values representing those levels (normally \(\zeta_1=0, \zeta_2=N_z-1\) for a grid of \(N_z\) vertical layers [excluding ghost layers]), and \(\zeta_*\) is the \(\zeta\) value of the inflection point of the sinh function.
The \(z\) coordinate and \(\zeta\) value of the inflection point are related via:
which can be inverted to:
2.5.2.2.2. General profile:
For a general monotonic function \(\psi()\) instead of sinh:
and the reference point \(\zeta_*\) is found by inverting:
numerically.
2.5.2.2.3. Duct flow:
The profile function grid_function='duct' generates a grid profile
for turbulent flow in ducts.
For a duct flow, most gradients are steepest close to the walls, requiring
very fine resolution near the walls.
We follow the method of Kim (1987) and use a Chebyshev-type grid with a cosine distribution of the grid points such that in the y direction:
where
and \(h = L_y/2\) is the duct half-width.
Currently this method is adapted for ducts where x is the stream-wise direction, z is the span-wise direction, and the walls are at \(y=y_0\) and \(y=y_0+L_y\).
In order to have fine enough resolution, the first grid point should be a distance \(\delta = 0.05 \, l_w\) from the wall, where:
and \(\tau_w\) is the shear wall stress. This is accomplished by using at least:
grid points in the y-direction.
After rounding up to the next integer value, the truncated condition:
(where ceil(x) is the ceiling function) gives practically identical results.
2.5.2.2.4. Example:
To apply the sinh profile, you can set the following in start.in
(this example is from samples/sound-spherical-noequi/):
&init_pars
[...]
xyz0 = -2., -2., -2. ! first corner of box
Lxyz = 4., 4., 4. ! box size
lperi = F , F , F ! periodic direction?
lequidist = F, F, T ! non-equidistant grid in z
xyz_star = , , -2. ! position of inflection point
grid_func = , , 'sinh' ! sinh function: linear for small, but
! exponential for large z
coeff_grid = , , 0.5
/
The parameter array coeff_grid represents \(z_*\) and \(a\); the bottom
height \(z_0\) and the total box height \(L_z\) are taken from xyz0 and Lxyz
as in the equidistant case.
The inflection point of the sinh profile (the part where it is linear) is
not in the middle of the box, because we have set xyz_star(3) (i.e. \(z_*\))
to -2.
2.5.3. Diagnostic output
Every it1 time steps (it1 is a runtime parameter, see
Sect. S-all-run-params), the code writes monitoring output to
stdout and, parallel to this, to the file data/time_series.dat.
The variables that appear in this listing and the output format are
defined in the file print.in and can be changed without touching
the code (even while it is running).
A simple example of print.in may look like this:
t(F10.3)
urms(E13.4)
rhom(F10.5)
oum
This means that the output table will contain:
Time
tin the first column, formatted asF10.3The mean squared velocity
urms`(i.e. \(\langle \mathbf{u}^2 \rangle^{1/2}\)) in the second column with formatE13.4The average density
rhom(i.e. \(\langle \rho \rangle\), which allows monitoring mass conservation) formattedF10.5The kinetic helicity
oum(i.e. \(\langle \vec{\omega} \cdot \mathbf{u} \rangle\)) in the last column with the default formatE10.2[3]
The corresponding diagnostic output will look like this:
----t---------urms--------rhom------oum----
0.000 4.9643E-03 14.42457 -8.62E-06
0.032 3.9423E-03 14.42446 -5.25E-06
0.063 6.8399E-03 14.42449 -3.50E-06
0.095 1.1437E-02 14.42455 -2.58E-06
0.126 1.6980E-02 14.42457 -1.93E-06
2.5.4. Data files
2.5.4.1. Snapshot files
Snapshot files contain the values of all evolving variables and are sufficient to restart a run. In the case of an MHD simulation with entropy equation, for example, the snapshot files will contain the values of velocity, logarithmic density, entropy and the magnetic vector potential.
There are two kinds of snapshot files: the current snapshot and permanent
snapshots, both of which reside in the directory data/proc$N$/.
The parameter isav determines the frequency at which the current snapshot
data/proc$N$/var.dat is written. If you keep this frequency too high,
the code will spend a lot of time on I/O, in particular for large jobs; too low
a frequency makes it difficult to follow the evolution interactively during
test runs.
There is also the ialive parameter. Setting this to 1 or 10 gives an updated
timestep in the files data/proc*/alive.info. You can put
ialive=0 to turn this off to limit the I/O on your machine.
The permanent snapshots data/proc*/VAR$N$ are written every
dsnap time units. These files are numbered consecutively from N=1 upward and
for long runs they can occupy quite some disk space. On the other hand, if
after a run you realize that some additional quantity q would have been
important to print out, these files are the only way to reconstruct the time
evolution of q without re-running the code.
2.5.4.1.1. File structure
Snapshot files consist of the following Fortran records [4] :
variable vector \(f [mx × my × mz × nvar]\)
time \(t\) [1], coordinate vectors \(x\) [
mx], \(y\) [my], \(z\) [mz], grid spacings \(\delta x\) [1], \(\delta y\) [1], \(\delta z\) [1], shearing-box shift \(\Delta y\) [1]
All numbers (apart from the record markers) are single precision (4-byte) floating point numbers, unless you use double precision (see Running in double-precision), in which case all numbers are 8-byte floating point numbers, while the record markers remain 4-byte integers.
The script pc_tsnap allows you to determine the time \(t\) of a snapshot file:
unix> pc_tsnap data/proc0/var.dat
data/proc0/var.dat: t = 8.32456
unix> pc_tsnap data/proc0/VAR2
data/proc0/VAR2: t = 2.00603
2.5.5. Video files and slices
We use the terms video files and slice files interchangeably. These files contain a time series of values of one variable in a given plane. The output frequency of these video snapshots is set by the parameter dvid (in code time units).
When output to video files is activated by some settings in
run.in (see example below) and the existence of video.in,
slices are written for four planes:
\(x\)-\(z\) plane (\(y\) index
iy; file suffix.xz)\(y\)-\(z\) plane (\(y\) index
ix; suffix.yz)\(x\)-\(y\) plane (\(y\) index
iz; suffix.xy)another slice parallel to the \(x\)-\(y\) plane (\(y\) index
iz2; suffix.xy2)
You can specify the position of the slice planes by setting the parameters
ix, iy, iz, and iz2 in the namelist run_pars
in run.in. Alternatively, you can set the input parameter
slice_position to one of 'p' (periphery of box) or 'm'
(middle of box). Or you can also specify the \(z\)-position using the
tags zbot_slice and ztop_slice. In this case, the
zbot_slice slice will have suffix .xy and ztop_slice the
suffix .xy2.
In the file video.in of your run directory, you can choose
for which variables you want to get video files; valid choices are listed
in S-video.in-params.
The slice files are written in each processor directory
data/proc*/ and have a file name indicating the individual
variable (e.g., slice_uu1.yz for a slice of \(u_x\) in
the \(y\)-\(z\) plane). Before visualizing slices one normally
wants to combine the sub-slices written by each processor into one global
slice (for each plane and variable). This is done by running
src/read_videofiles.x, which will prompt for the variable
name, read the individual sub-slices and write global slices to
data/. Once all global slices have been assembled you may
want to remove the local slices data/proc*/slice*.
To read all sub-slices demanded in video.in at once, use
src/read_all_videofiles.x. This program doesn’t expect any
user input and can thus be submitted in computing queues.
For visualization of slices, you can use the IDL routines
rvid_box.pro, rvid_plane.pro, or rvid_line.pro,
which allow the flag /png for writing PNG images that can then be
combined into an MPEG movie using mpeg_encode. Based on
rvid_box, you can write your own video routines in IDL.
2.5.5.1. An example
Suppose you have set up a run using entropy.f90 and magnetic.f90
(most probably together with hydro.f90 and other modules). In order
to animate slices of entropy \(s\) and the \(z\)-component
\(B_z\) of the magnetic field, in planes passing through the center
of the box, do the following:
Write the following lines to
video.inin your run directory:ss bb divu
Edit the namelist run_pars in the file
run.in. Request slices by settingwrite_slicesand setdvidandslice_positionto reasonable values, e.g.:!lwrite_slices=T !(no longer works; write requested slices into video.in) dvid=0.05 slice_position='m'
Run the Pencil Code:
$ start.csh $ run.csh
Say
make read_videofilesto compile the routine and then runsrc/read_videofiles.xto assemble global slice files from those scattered acrossdata/proc*/:$ src/read_videofiles.x enter name of variable (lnrho, uu1, ..., bb3): ss $ src/read_videofiles.x enter name of variable (lnrho, uu1, ..., bb3): bb3
Start IDL and run
rvid_box:$ idl IDL> rvid_box,'bb3' IDL> rvid_box,'ss',min=-0.3,max=2. etc.
2.5.5.1.1. Another example
Suppose you have set up a run using magnetic.f90 and some other modules.
This run studies some process in a surface layer inside the box. This
surface can represent a sharp change in density or turbulence. So you defined
your box setting the \(z=0\) point at the surface.
Therefore, your start.in file will look something similar to:
&init_pars
lperi=T,T,F
bcz = 's','s','a','hs','s','s','a'
xyz0 = -3.14159, -3.14159, -3.14159
Lxyz = 6.28319, 6.28319, 9.42478
A smarter way of specifying the box size in units of \(\pi\) is to write
&init_pars
xyz_units = 'pi', 'pi', 'pi'
xyz0 = -1., -1., -1.
Lxyz = 2., 2., 2.
Now you can analyze quickly the surface of interest and some other \(xy\) slice
setting zbot_slice and ztop_slice in the run.in file:
&run_pars
slice_position='c'
zbot_slice=0.
ztop_slice=0.2
In this case, the slices with the suffix .xy will be at the surface
and the ones with the suffix .xy2 will be at the position \(z=0.2\) above
the surface. And you can visualize this slices by:
Write the following lines to
video.inin your run directory:bb
Edit the namelist
run_parsin the filerun.into includezbot_sliceandztop_slice.Run the Pencil Code:
unix> start.csh unix> run.csh
Run src/read_videofiles.x to assemble global slice files from those scattered across
data/proc*/:unix> src/read_videofiles.x enter name of variable (lnrho, uu1, ..., bb3): bb3
Start IDL, load the slices with
pc_read_videoand plot them at some time:unix> idl IDL> pc_read_video, field='bb3',ob=bb3,nt=ntv IDL> tvscl,bb3.xy(*,*,100) IDL> tvscl,bb3.xy2(*,*,100) etc.
2.5.5.1.2. File structure
Slice files consist of one Fortran record [5] for each slice,
which contains the data of the variable (without ghost zones), the time
\(t\) of the snapshot and the position of the sliced variable
(e.g., the \(x\) position for a \(y\)-\(z\) slice):
data \(_1\) [\(nx \times ny \times nz\)], time \(t_1\) [1], position \(_1\) [1]
data \(_2\) [\(nx \times ny \times nz\)], time \(t_2\) [1], position \(_2\) [1]
data \(_3\) [\(nx \times ny \times nz\)], time \(t_3\) [1], position \(_3\) [1]
etc.
2.5.6. Averages
2.5.6.1. One-dimensional output averaged in two dimensions
In the file xyaver.in, \(z\)-dependent (horizontal) averages
are listed. They are written to the file data/xyaverages.dat. A
new line of averages is written every it1 th time steps.
There is the possibility to output two-dimensional averages. The result
then depends on the remaining dimension. The averages are listed in the
files xyaver.in, xzaver.in, and yzaver.in where
the first letters indicate the averaging directions. The output is then
stored to the files data/xyaverages.dat, data/xzaverages.dat,
and data/yzaverages.dat. The output is written every it1d
time steps.
The rms values of the so defined mean magnetic fields are referred to as
bmz, bmy and bmx, respectively, and the rms values
of the so defined mean velocity fields are referred to as umz,
umy, and umx. (The last letter indicates the direction on
which the averaged quantity still depends.)
See Adding new output diagnostics on how to add new averages.
In idl such \(xy\)-averages can be read using the procedure
pc_read_xyaver.
2.5.6.2. Two-dimensional output averaged in one dimension
There is the possibility to output one-dimensional averages. The result
then depends on the remaining two dimensions. The averages are listed in
the files yaver.in, zaver.in, and phiaver.in where
the first letter indicates the averaging direction. The output is then
stored to the files data/yaverages.dat, data/zaverages.dat,
and data/phiaverages.dat.
See Adding new output diagnostics on how to add new averages.
Disadvantage
The output files, e.g., data/zaverages.dat, can be rather big because each average is just appended to the file.
2.5.6.3. Azimuthal averages
Azimuthal averages are controlled by the file phiaver.in, which
currently supports the quantities listed in S-phiaver.in-params.
In addition, one needs to set lwrite_phiaverages, lwrite_yaverages,
or lwrite_zaverages to \(.true.\). For example, if
phiaver.in contains the single line:
b2mphi
then you will get azimuthal averages of the squared magnetic field \(\Bv^2\).
Azimuthal averages are written every d2davg time units to the
files data/averages/PHIAVG$N$. The file format of azimuthal-average
files consists of the following Fortran records [6]:
number of radial points \(N_{r,\rm \phi-avg}\) [1], number of vertical points \(N_{z,\rm \phi-avg}\) [1], number of variables \(N_{\rm var,\phi-avg}\) [1], number of processors in \(z\) direction [1]
time \(t\) [1], positions of cylindrical radius \(r_{\rm cyl}\) [\(N_{r,\rm \phi-avg}\)] and \(z\) [\(N_{z,\rm \phi-avg}\)] for the grid, radial spacing \(\delta r_{\rm cyl}\) [1], vertical spacing \(\delta z\) [1]
averaged data [\(N_{r,\rm \phi-avg} {\times} N_{z,\rm \phi-avg}\)]
label length [1], labels of averaged variables [\(N_{\rm var,\phi-avg}\)]
All numbers are 4-byte numbers (floating-point numbers or integers), unless you use double precision (see Running in double-precision).
To read and visualize azimuthal averages in IDL, use
$PENCIL_HOME/idl/files/pc_read_phiavg.pro:
IDL> avg = pc_read_phiavg('data/averages/PHIAVG1')
IDL> contour, avg.b2mphi, avg.rcyl, avg.z, TITLE='!17B!U2!N!X'
or have a look at $PENCIL_HOME/idl/phiavg.pro for a more sophisticated example.
2.5.6.4. Time averages
Time averages need to be prepared in the file src/ctimeavg.local,
since they use extra memory. They are controlled by the averaging time
\(\tau_{\rm avg}\) (set by the parameter tavg in run.in),
and by the indices idx_tavg of variables to average.
Currently, averaging is implemented as exponential (memory-less) average [7] :
which is equivalent to
Here \(t_0\) is the time of the snapshot the calculation started with, i.e., the snapshot read by the last run.x command. Note that the implementation will approximate the integral only to first-order accuracy in \(\delta t\). In practice, however, \(\delta t\) is small enough to make this accuracy suffice.
In src/ctimeavg.local, you need to set the number of slots used for
time averages. Each of these slots uses \(\mathtt{mx}\times\mathtt{my}\times\mathtt{mz}\)
floating-point numbers, i.e., half as much memory as each fundamental variable.
For example, if you want to get time averages of all variables, set:
integer, parameter :: mtavg=mvar
in src/ctimeavg.local, and don’t set idx_tavg in run.in.
If you are only interested in averages of variables 1–3 and 6–8 (say,
the velocity vector and the magnetic vector potential in a run with
hydro.f90, density.f90, entropy.f90 and
magnetic.f90), then set:
integer, parameter :: mtavg=6
in src/ctimeavg.local, and set:
idx_tavg = 1,2,3,6,7,8 ! time-average velocity and vector potential
in run.in.
Permanent snapshots of time averages are written every tavg time
units to the files data/proc*/TAV$N$. The current time averages
are saved periodically in data/proc*/timeavg.dat whenever
data/proc*/var.dat is written. The file format for time averages
is equivalent to that of the snapshots; see Snapshot files above.
2.5.7. Helper scripts
The bin directory contains a collection of utility scripts,
some of which are discussed elsewhere. Here is a list of the more important ones.
adapt-mkfileActivate the settings in a
Makefilethat apply to the given computer, see Adapting Makefile.src [obsolete; see Sect.:ref:man1_configuration].auto-testVerify that the code compiles and runs in a set of run directories and compare the results to the reference output. These tests are carried out routinely to ensure that the svn version of the code is in a usable state.
cleanf95Can be use to clean up the output from the Intel x86 Fortran 95 compiler (ifc).
copy-proc-to-procUsed for restarting in a different directory. Example:
copy-proc-to-proc seed.dat ../hydro256ecopy-snapshotsCopy snapshots from a processor-local directory to the global directory. To be started in the background before
run.xis invoked. Used bystart.cshandrun.cshon network connected processors.pc_copyvar var1 var2 source destCopies snapshot files from one directory (source) to another (dest). See documentation in file.
pc_copyvar v v dirCopies all
var.datfiles from current directory tovar.datindirrun directory. Used for restarting in a different directory.pc_copyvar N vUsed to restart a run from a particular snapshot
VAR$N$. Copies a specified snapshotVAR$N$tovar.datwhereNand (optionally) the target run directory are given on the command line.cvs-add-rundirAdd the current run directory to the svn repository.
cvsci_runSimilar to
cvs-add-rundir, but it also checks in the*.inandsrc/*.localfiles. It also checks in the filesdata/time_series.dat,data/dim.datanddata/index.profor subsequent processing in IDL on another machine. This is particularly useful if collaborators want to check each others’ runs.dx_*These script perform several data collection or reformatting exercises required to read particular files into DX. They are called internally by some of the DX macros in the
dx/macrosdirectory.getconf.cshSee start.csh, run.csh, getconf.csh` [obsolete; see Sect. man1_configuration]
gpgrowthPlot simple time evolution with Gnuplot’s ASCII graphics for fast orientation via a slow modem line.
localMaterialize a symbolic link.
mkcparamBased on
MakefileandMakefile.local, generatesrc/cparam.inc, which specifies the numbermvarof fundamental variables, andmauxof auxiliary variables. Called by theMakefile.pc_mkdatadirCreates a link to a data directory in a suitable workspace. By default this is on
/var/tmp/, but different locations are specified for different machines.mkdotinGenerate minimal
start.in,run.infiles based onMakefileandMakefile.local.mkinparsWrapper around
mkdotin— needs proper documentation.mkproc-treeGenerates a multi-processor (
proc$N$/) directory structure. Useful when copying data files in a processor tree, such as slice files.mkwwwGenerates a template HTML file for describing a run of the code, showing input parameters and results.
move-sliceMoves all the slice files from a processor tree structure,
proc$N$/, to a new target tree creating directories where necessary.nl2idlTransform a Fortran namelist (normally the files
param.nml,param2.nmlwritten by the code) into an IDL structure. Generates an IDL file that can be sourced fromstart.proorrun.pro.pacx-adapt-makefileVersion of adapt-makefile for highly distributed runs using PACX MPI.
pc_newrunGenerates a new run directory from an old one. The new one contains a copy of the old
*.localfiles, runspc_setupsrc, and makes also a copy of the old*.inandk.datfiles.pc_newscanGenerates a new scan directory from an old one. The new one contains a copy of the old, e.g., the one given under
samples/parameter_scan. Look in theREADMEfile for details.pc_inspectrunCheck the execution of the current run: prints legend and the last few lines of the
time_series.datfile. It also appends this result to a file calledSPEED, which contains also the current wall clock time, so you can work out the speed of the code (without being affected by i/o time).read_videofiles.cshThe script for running read_videofiles.x.
remote-topCreate a file
top.login the relevantproc$N$directory containing the output oftopfor the appropriate processor. Used in batch scripts for multi-processor runs.run.cshThe script for producing restart files with the initial condition; see start.csh, run.csh, getconf.csh` [obsolete; see Sect. man1_configuration]
scpdatadirMake a tarball of data directory,
data/and usescpto secure copy to copy it to the specified destination.pc_setupsrcLink
start.csh,run.cshandgetconf.cshfrom$PENCIL_HOME/bin. Generatesrc/if necessary and link the source code files from$PENCIL_HOME/srcto that directory.start.cshThe script for initializing the code; see start.csh, run.csh, getconf.csh` [obsolete; see Sect. man1_configuration]
summarize-historyEvaluate
params.logand print a history of changes.timestrGenerate a unique time string that can be appended to file names from shell scripts through the backtick mechanism.
pc_tsnapExtract time information from a snapshot file,
VAR$N$.
There are several additional scripts on pencil-code/utils. Some are located
in separate folders according to users. There could be redundancies, but it is often
just as easy to write your own new script than figuring out how something else works.
2.5.8. RELOAD, STOP and SAVE files
The code periodically (every it time steps) checks
for the existence of two files, RELOAD
and STOP, which can be used to trigger certain behavior.
2.5.8.1. Reloading run parameters
In the directory where you started the code, create the file
RELOAD with
touch RELOAD
to force the code to re-read the runtime parameters from run.in.
This will happen the next time the code is writing monitoring output (the
frequency of this happening is controlled by the input parameter it,
see Start and run parameters).
Each time the parameters are reloaded, the new set of parameters is
appended (in the form of namelists) to the file
data/params.log together with the time \(t\), so you have
a full record of your changes.
If RELOAD contains any text, its first line will be written to
data/params.log as well, which allows you to annotate
changes:
echo "Reduced eta to get fields growing" > RELOAD
Use the command summarize-history to print a history of changes.
2.5.8.2. Stopping the code
In the directory where you started the code, create the file
STOP with
touch STOP
to stop the code in a controlled manner (it will write the latest snapshot). Again, the action will happen the next time the code is writing monitoring output.
2.5.8.3. Saving a snapshot
In the directory where you started the code, create the file
SAVE with
touch SAVE
to save the current state of the simulation in the file var.dat.
See Stopping the code for when this action is taken.
2.5.9. RERUN and NEWDIR files
After the code finishes (e.g., when the final timestep number is reached
or when a STOP file is found), the run.csh script checks
whether there is a RERUN file.
If so, the code will simply run again, perhaps even after you have
recompiled the code.
This is useful in the development phase when you changed something in
the code, so you don’t need to wait for a new slot in the queue!
Even more naughty, as Tony says, is the NEWDIR file, where
you can enter a new directory path (relative path is ok, e.g.,
../conv-slab).
If nothing is written in this file (e.g., via touch NEWDIR)
it stays in the same directory.
On distributed machines, the NEWDIR method will copy all the
VAR# and var.dat files back to and from the sever.
This can be useful if you want to run with new data files, but you
better do it in a separate directory, because with NEWDIR
the latest data from the code are written back to the server before
running again.
Oh, by the way, if you want to be sure that you haven’t messed up the
content of the pair of NEWDIR files, you may want to try out
the pc_jobtransfer command.
It writes the decisive STOP file only after the script has
checked that the content of the two NEWDIR files points to
existing run directory paths, so if the new run crashes, the code
returns safely to the old run directory.
2.5.10. Start and run parameters
All input parameters in start.in and run.in are grouped in
Fortran namelists.
This allows arbitrary order of the parameters (within the given
namelist; the namelists need no longer be in the correct order), as well as
enhanced readability through inserted Fortran comments and whitespace.
One namelist (init_pars / run_pars) contains general
parameters for initialization/running and is always read in.
All other namelists are specific to individual modules and will only
be read if the corresponding module is used.
The syntax of a namelist (in an input file like start.in) is
&init_pars
ip=5, Lxyz=2,4,2
/
— in this example, the name of the namelist is init_pars, and we
read just two variables (all other variables in
the namelist retain their previous value): ip, which is set to \(5\),
and Lxyz, which is a vector of length three and is set to \((2,4,2)\).
While all parameters from the namelists can be set, in most cases
reasonable default values are preset.
Thus, the typical file start.in will only contain a minimum set of
variables or (if you are very minimalistic) none at all.
If you want to run a particular problem, it is best to start by
modifying an existing example that is close to your application.
Before starting a simulation run, you may want to execute the command pc_configtest
in order to test the correctness of your changes to these configuration files.
As an example, we give here the start parameters for
samples/helical-MHDturb/
&init_pars
cvsid='${}Id:$', ! identify version of start.in
xyz0 = -3.1416, -3.1416, -3.1416, ! first corner of box
Lxyz = 6.2832, 6.2832, 6.2832, ! box size
lperi = T , T , T , ! periodic in x, y, z
random_gen='nr_f90'
/
&hydro_init_pars
/
&density_init_pars
gamma=1.
/
&magnetic_init_pars
initaa='gaussian-noise', amplaa=1e-4
/
The three entries specifying the location, size and periodicity of the box are just given for demonstration purposes here — in fact a periodic box from \(-\pi\) to \(-\pi\) in all three directions is the default. In this run, for reproducibility, we use a random number generator from the Numerical Recipes [NR], rather than the compiler’s built-in generator. The adiabatic index \(\gamma\) is set explicitly to \(1\) (the default would have been 5/3) to achieve an isothermal equation of state. The magnetic vector potential is initialized with uncorrelated, normally distributed random noise of amplitude \(10^{-4}\).
The run parameters for samples/helical-MHDturb/ are
&run_pars
cvsid='${}Id:$', ! identify version of start.in
nt=10, it1=2, cdt=0.4, cdtv=0.80, isave=10, itorder=3
dsnap=50, dvid=0.5
random_gen='nr_f90'
/
&hydro_run_pars
/
&density_run_pars
/
&forcing_run_pars
iforce='helical', force=0.07, relhel=1.
/
&magnetic_run_pars
eta=5e-3
/
&viscosity_run_pars
nu=5e-3
/
Here we run for nt \(=10\) timesteps, every second step, we write a
line of diagnostic output; we require the time step to keep the advective
Courant number \(\le 0.4\) and the diffusive Courant number
\(\le 0.8\), save var.dat every 20 time steps, and
use the 3-step time-stepping scheme described in Appendix S-2N-scheme
(the Euler scheme itorder \(=1\) is only useful for tests).
We write permanent snapshot file VAR N every dsnap \(=50\) time
units and 2d slices for animation every dvid \(=0.5\) time units.
Again, we use a deterministic random number generator.
Viscosity \(\nu\) and magnetic diffusivity \(\eta\)
are set to \(5\times10^{-3}\) (so the mesh Reynolds number
based on the rms velocity Mesh Reynolds number is about
\(u_{\rm rms}\delta x/\nu=0.3\times(2\pi/32)/5\times10^{-3}\approx12\),
which is in fact rather a bit to high).
The parameters in forcing_run_pars specify fully helical forcing of
a certain amplitude.
A full list of input parameters is given in Appendix S-all-parameters.
2.5.11. Physical units
Many calculations are unit-agnostic, in the sense that all results remain the same independent of the unit system in which you interpret the numbers. E.g. if you simulate a simple hydrodynamical flow in a box of length \(L=1.\) and get a maximum velocity of \(u_{\rm max}=0.5\) after \(t=3\) time units, then you may interpret this as \(L=1 {\rm m}\), \(u_{\rm max}= 0.5 {\rm m/s}\), \(t= 3 {\rm s}\), or as \(L=1 {\rm pc}\), \(u_{\rm max}= 0.5 {\rm pc/Myr}\), \(t= 3 {\rm Myr}\), depending on the physical system you have in mind. The units you are using must of course be consistent, thus in the second example above, the units for diffusivities would be \({\rm pc^2/Myr}\), etc.
The units of magnetic field and temperature are determined by the values \(\mu_0=1\) and \(c_p=1\) used internally by the code [8] . This means that if your units for density and velocity are \([\rho]\) and \([v]\), then magnetic fields will be in
and temperatures are in
\([\rho]\) |
\([v]\) |
\([B]\) |
\([T]\) |
|---|---|---|---|
1 kg/m \(^3\) |
1 m/s |
1.12 mT = 11.2 G |
\(\left(\dfrac{\bar{\mu}_{\rm g}}{0.6}\right) \times 2.89\EE{-5}\, {\rm K}\) |
1 g/cm \(^3\) |
1 cm/s |
3.54 \(\EE{-4}\) T = 3.54 G |
\(\left(\dfrac{\bar{\mu}_{\rm g}}{0.6}\right) \times 2.89\, {\rm nK}\) |
1 g/cm \(^3\) |
1 km/s |
35.4 T = 354 kG |
\(\left(\dfrac{\bar{\mu}_{\rm g}}{0.6}\right) \times 28.9\, {\rm K}\) |
1 g/cm \(^3\) |
10 km/s |
354 T = 3.54 MG |
\(\left(\dfrac{\bar{\mu}_{\rm g}}{0.6}\right) \times 2\,890\, {\rm K}\) |
For some choices of density and velocity units, Table 2.2 shows the resulting units of magnetic field and temperature.
On the other hand, as soon as material equations are used (e.g., one of
the popular parameterizations for radiative losses, Kramers opacity,
Spitzer conductivities or ionization, which implies well-defined
ionization energies), the corresponding routines in the code need to know
the units you are using.
This information is specified in start.in or run.in through
the parameters unit_system,
unit_length, unit_velocity, unit_density and
unit_temperature [9] like e.g.
unit_system='SI',
unit_length=3.09e16, unit_velocity=978. ! [l]=1pc, [v]=1pc/Myr
Note
The default unit system is unit_system='cgs' which is a synonym for unit_system='Babylonian cubits'.
2.5.12. Minimum amount of viscosity
We emphasize that, by default, the code works with constant diffusion coefficients (viscosity \(\nu\), thermal diffusivity \(\chi\), magnetic diffusivity \(\eta\), or passive scalar diffusivity \(\mathcal{D}\)). If any of these numbers is too small, you would need to have more meshpoints to get consistent numerical solutions; otherwise the code develops wiggles (ringing’) and will eventually crash. A useful criterion is given by the mesh Reynolds number *based on the maximum velocity* :index:`Mesh Reynolds number,
which should not exceed a certain value which can be problem-dependent. Often the largest possible value of \(\mbox{Re}_{\rm mesh}\) is around 5. Similarly there exist mesh Péclet and mesh magnetic Reynolds numbers that should not be too large.
Note that in some cases, wiggles’ in :math:lnrho` will develop despite sufficiently large diffusion coefficients, essentially because the continuity equation contains no dissipative term. For convection runs (but not only for these), we have found that this can often be prevented by Upwinding, see Sect.~ref{S-upwind}.
If the Mach number of the code approaches unity, i.e. if the rms velocity becomes comparable with the speed of sound, shocks may form. In such a case the mesh Reynolds number should be smaller. In order to avoid excessive viscosity in the unshocked regions, one can use the so-called shock viscosity (Sect.~ref{ShockViscosity}) to concentrate the effects of a low mesh Reynolds number to only those areas where it is necessary.
2.5.13. The time step
2.5.13.1. The usual RK-2N time step
RK-2N refers to the third order Runge-Kutta scheme by Williamson (1980) with a memory consumption of two chunks. Therefore the 2N in the name.
The time step is normally specified as Courant time step through the coefficients cdt (\(c_{\delta t}\)), cdtv (\(c_{\delta t,{\rm v}}\)) and cdts (\(c_{\delta t,{\rm s}}\)). The resulting Courant step is given by
where
\(\cs\) and \(\vA\) denoting sound speed and Alfvén speed, respectively;
where \(\nu\) denotes kinematic viscosity, \(\chi = K/(c_p\rho)\) thermal diffusivity and \(\eta\) the magnetic diffusivity;
where dots indicate the presence of other terms on the rhs of the entropy equation.
To fix the time step \(\delta t\) to a value independent of velocities and
diffusivities, explicitly set the run parameter dt, rather than
cdt or cdtv (see p.~:ref:dt-run).
If the time step exceeds the viscous time step the simulation may actually run ok for quite some time. Inspection of images usually helps to recognize the problem. An example is shown in Fig. 2.3.
Fig. 2.3 Example of a velocity slice from a run where the time step is too long. Note the spurious checkerboard modulation in places, for example near \(x=-0.5\) and \(-2.5<y<-1.5\). This is an example of a hyperviscous turbulence simulations with \(512^3\) meshpoints and a third order hyperviscosity of \(\nu_3=5\times10^{-12}\). Hyperviscosity is explained in the Appendix~:ref:high-freq-filters.
Timestepping is accomplished using the Runge-Kutta 2N scheme. Regarding details of this scheme see Sect.~:ref:S-2N-scheme.
2.5.13.2. The Runge-Kutta-Fehlberg time step
A fifth order Runge-Kutta-Fehlberg time stepping procedure is available. It is used mostly for chemistry application, often together with the double precision option. In order to make this work, you need to compile with
TIMESTEP = timestep_rkf
in src/Makefile.local.
In addition, you must put itorder=5 in run.in.
An example application is
samples/1d-tests/H2_flamespeed (in H2_flamespeed/).
Attention
This procedure is still experimental.
2.5.14. Boundary conditions
2.5.14.1. Where to specify boundary conditions
In most tests that come with the PENCIL CODE, boundary conditions are set
in run.in, which is a natural choice.
However, this may lead to unexpected initial data written by
start.x, since when you start the code (via start.csh), the
boundary conditions are unknown and start.x will then fill the ghost
zones assuming periodicity (the default boundary condition) in all three
directions.
These ghost data will never be used in a calculation, as run.x will
apply the boundary conditions before using any ghost-zone values.
To avoid these periodic conditions in the initial snapshot, you
can set the boundary conditions in start.in already.
In this case, they will be inherited by run.x, unless you also
explicitly set boundary conditions in run.in.
2.5.14.2. How to specify boundary conditions
Boundary conditions are implemented through three layers of
ghost points on either boundary, which is quite a natural choice
for an MPI code that uses ghost zones for representing values located on
the neighboring processors anyway.
The desired type of boundary condition is set through the parameters
bc{x,y,z}
in run.in; the nomenclature used is as follows.
Set bc{x,y,z} to a sequence of letters like
bcx = 'p','p','p', 'p', 'p'
for periodic boundaries, or
bcz = 's','s','a','a2','c1:c2'
for non-periodic ones. Each element corresponds to one of the variables, which are those of the variables \(u_x\), \(u_y\), \(u_z\), \(\ln\rho\), \(s/c_p\), \(A_x\), \(A_y\), \(A_z\), \(\ln c\) that are actually used in this order. The following conditions are available:
p – periodic boundary condition
a – antisymmetric condition w.r.t. the boundary (vanishing value)
s – symmetric condition w.r.t. the boundary (vanishing first derivative)
a2 – antisymmetry w.r.t. the arbitrary value on the boundary (vanishing second derivative)
c1 – constant heat flux through the boundary (for \(\ln\rho\) and \(s\))
c2 – constant temperature at the boundary (for \(s\); requires
a2for \(\ln\rho\))cT – constant temperature at the boundary (for arbitrarily set \(\ln\rho\))
ce – set ghost-point temperature equal to boundary value (for arbitrarily set \(\ln\rho\))
db – low-order one-sided derivatives (“no boundary condition”) for density
she – shearing-sheet boundary condition (default when Shear is used)
g – force boundary field values (requires force_lower_bound and force_upper_bound in
init_pars)hs – enforce hydrostatic equilibrium at vertical boundaries (for \(\ln\rho\) and \(s\))
The extended syntax a:b (e.g. c1:c2) means: use boundary condition a at the lower boundary, but b at the upper one.
If you build a new run.in from another one with a different number
of variables (noentropy vs. entropy, for example), you must
adjust the length of the arrays bcx to bcz.
The advantage of this approach is that it is easy to replace all boundary conditions
in one direction at once (e.g. make everything periodic, or switch off shearing-sheet
conditions and use stress-free boundaries instead).
2.5.15. Restarting a simulation
When a run stops at the end of a simulation, you can just resubmit
the job again, and it will start from the latest snapshot saved in
data/proc*/var.dat. The value of the latest time is saved in a
separate file, data/proc*/time.dat.
On parallel machines it is possible that some (or just one) of the
var.dat are corrupt; for example after a system crash.
Check for file size and date, and restart from a good VARN
file instead.
If you want to run on a different machine, you just need to copy the
data/proc*/var.dat (and, just to be sure)
data/proc*/time.dat) files into a new directory tree.
You may also need the data/proc*/seed.dat
files for the random number generator. The easiest way to get all these
other files is to run start.csh again on the new machine (or in
a new directory) and then to overwrite the
data/proc*/var.dat files with the correct ones.
For restarting from runs that didn’t have magnetic fields, passive scalar
fields, or test fields, see Sect. RestartingFromLessPhysics.
2.5.16. One- and two-dimensional runs
If you want to run two-dimensional problems, set the number
of mesh points in one direction to unity, e.g., nygrid`=1
or :file:`nzgrid`=1 in :file:`cparam.local.
Remember that the number of mesh points is still divisible by
the number of processors.
For 2D-runs, it is also possible to write only 2D-snapshots (i.e. VAR
files written only in the considered (\(x\),:math:y) or (\(x\),:math:z) plane, with a size
seven times smaller as we do not write the third unused direction). To
do that, please add the logical flag lwrite_2d=T in the namelist init_pars in start.in.
Similarly, for one-dimensional problems, set, for example,
nygrid=1 and nzgrid=1 in cparam.local.
You can even do a zero-dimensional run, but then you better
set dt (rather than cdt), because there is no
Courant condition for the time step.
See 0d, 1d, 2d, and 3d tests with examples.
2.5.17. Visualization
2.5.17.1. Gnuplot
Simple visualization can easily be done using Gnuplot, an open-source plotting program suitable for two-dimensional plots.
For example, suppose you have the variables
---it-----t-------dt-------urms-----umax-----rhom-----ssm------dtc---
in time_series.dat and want to plot \(u_{\rm rms}(t)\).
Just start gnuplot and type
gnuplot> plot "data/time_series.dat" using 2:4 with lines
If you work over a slow line and want to see both \(u_{\rm rms}(t)\) and \(u_{\rm max}(t)\), use ASCII graphics:
gnuplot> set term dump
gnuplot> set logscale y
gnuplot> plot "data/time_series.dat" using 2:4 title "urms", \
gnuplot> "data/time_series.dat" using 2:5 title "umax"
2.5.17.2. Data explorer
DX (data explorer; http://www.opendx.org) is an open-source tool for visualization of three-dimensional data.
PENCIL CODE provides a few networks for DX. It is quite easy to read in a snapshot file from DX (the only tricky thing is the four extra bytes at the beginning of the file, representing a Fortran record marker), and whenever you run start.x, the code writes a file var.general that tells DX all it needs to know about the data structure.
As a starting point for developing your own DX programs or networks, you can use a few generic DX scripts provided in the directory dx/basic/.
From the run directory, start DX with
unix> dx -edit $PENCIL_HOME/dx/basic/lnrho
to load the file dx/basic/lnrho.net, and execute it with Ctl-o or Execute -> Execute Once.
You will see a set of iso-surfaces of logarithmic density.
If the viewport does not fit to your data, you can reset it with Ctl-f.
To rotate the object, drag the mouse over the Image window with the left or right mouse button pressed.
Similar networks are provided for entropy (ss.net), velocity (uu.net) and magnetic field (bb.net).
When you expand these simple networks to much more elaborate ones, it is probably a good idea to separate the different tasks (like Importing and Selecting, visualizing velocity, visualizing entropy, and Rendering) onto separate pages through Edit -> Page.
Note
Currently, DX can only read in data files written by one single processor, so from a multi-processor run, you can only visualize one subregion at a time.
2.5.17.3. GDL
GDL, also known as Gnu Data Language is a free visualization package that can be found at http://gnudatalanguage.sourceforge.net/. It aims at replacing the very expensive IDL package (see IDL). For the way we use IDL for PENCIL CODE, compatibility is currently not completely sufficient, but you can use GDL for many of the visualization tasks. If you get spurious “Error opening file” messages, you can normally simply ignore them.
2.5.17.3.1. Setup
As of GDL 0.9 – at least the version packed with Ubuntu Jaunty (9.10) – you will need to add GDL’s examples/pro/ directory to your !PATH variable.
So the first call after starting GDL should be
GDL> .run setup_gdl
2.5.17.3.2. Starting visualization
There are mainly two possibilities for visualization: using a simple GUI or loading the data with pc_read and work with it interactively. Please note that the GUI was written and tested only with IDL (see Visualization in IDL).
Here, the pc_read family of IDL routines to read the data is described. Try
GDL> pc_read
to get an overview.
To plot a time series, use
GDL> pc_read_ts, OBJECT=ts
GDL> help, ts, /STRUCT ;; (to see which slots are available)
GDL> plot, ts.t, ts.umax
GDL> oplot, ts.t, ts.urms
Alternatively, you could simply use the ts.pro script:
GDL> .run ts
To work with data from var.dat and similar snapshot files, you can e.g., use the following routines:
GDL> pc_read_dim, OBJECT=dim
GDL> $$PENCIL_HOME/bin/nl2idl -d ./data/param.nml> ./param.pro
GDL> pc_read_param, OBJECT=par
GDL> pc_read_grid, OBJECT=grid
GDL> pc_read_var, OBJECT=var
Having thus read the data structures, we can have a look at them to see what information is available:
GDL> help, dim, /STRUCT
GDL> help, par, /STRUCT
GDL> help, grid, /STRUCT
GDL> help, var, /STRUCT
To visualize data, we can e.g., do [10] :
GDL> plot, grid.x, var.ss[*, dim.ny/2, dim.nz/2]
GDL> contourfill, var.ss[*, *, dim.nz/2], grid.x, grid.y
GDL> ux_slice = var.uu[*, *, dim.nz/2, 0]
GDL> uy_slice = var.uu[*, *, dim.nz/2, 1]
GDL> wdvelovect, ux_slice, uy_slice, grid.x, grid.y
GDL> surface, var.lnrho[*, *, dim.nz/2, 0]
See also Sect. IDL.
2.5.17.4. IDL
IDL is a commercial visualization program for two-dimensional and simple three-dimensional graphics. It allows to access and manipulate numerical data in a fashion quite similar to how Fortran handles them.
In $PENCIL_HOME/idl we provide a number of general-purpose IDL scripts that we are using all the time in connection with PENCIL CODE.
While IDL is quite an expensive software package, it is quite useful for visualizing results from numerical simulations.
In fact, for many applications, the 7-minute demo version of IDL is sufficient.
2.5.17.4.1. Visualization in IDL
The Pencil Code GUI is a data post-processing tool for the usage on a day-to-day basis. It allows fast inspection of many physical quantities, as well as advanced features like horizontal averages, streamline tracing, freely orientable 2D-slices, and extraction of cut images and movies. To use the Pencil Code GUI, it is sufficient to run:
unix> idl
IDL> .r pc_gui
If you like to load only some subvolume of the data, like any 2D-slices from the given data snapshots, or 3D-subvolumes, it is possible to choose the corresponding options in the file selector dialog. The Pencil Code GUI offers also some options to be set on the command-line, please refer to their description in the source code.
There are also other small GUIs available, e.g., the file time-series.dat can easily be analyzed with the command:
unix> idl
IDL> pc_show_ts
The easiest way to derive physical quantities at the command-line is to use one of the many pc_read_var-variants (pc_read_var_raw is recommended for large datasets because of its high efficiency regarding computation and memory usage) for reading the data. With that, one can make use of pc_get_quantity to derive any implemented physical quantity. Packed in a script, this is the recommended way to get reproducible results, without any chance for accidental errors on the interactive IDL command-line.
Alternatively, by using the command-line to see the time evolution of e.g., velocity and magnetic field (if they are present in your run), start IDL and run ts.pro:
unix> idl
IDL> .run ts
The IDL script ts.pro script reads the time series data from data/time_series.dat and sorts the column into the structure ts, with the slot names corresponding to the name of the variables (taken from the header line of time_series.dat).
Thus, you can refer to time as ts.t, to the rms velocity as ts.urms, and in order to plot the mean density as a function of time, you would simply type:
IDL> plot, ts.t, ts.rhom
The basic command sequence for working with a snapshot is:
unix> idl
IDL> .run start
IDL> .run r
IDL> [specific commands] \
You run start.pro once to initialize (or reinitialize, if the mesh size has changed, for example) the fields and read in the startup parameters from the code.
To read in a new snapshot, run r.pro (or rall.pro, see below).
If you are running in parallel on several processors, the data are scattered over different directories. To reassemble everything in IDL, use:
IDL> .r rall
instead of .r r (here, .r is a shorthand for .run).
The procedure rall.pro reads (and assembles) the data from all processors and correspondingly requires large amounts of memory for very large runs.
If you want to look at just the data from one processor, use r.pro instead.
If you need the magnetic field or the current density, you can calculate them in IDL by:
IDL> bb=curl(aa)
IDL> jj=curl2(aa)
By default one is reading always the current snapshot data/proc$N$/var.dat; if you want to read one of the permanent snapshots, use (for example):
IDL> varfile='VAR2'
IDL> .r r (or .r rall)
With r.pro, you can switch the part of the domain by changing the variable datadir:
IDL> datadir='data/proc3'
IDL> .r r
will read the data written by processor 3.
Note
If you run IDL from the command line, you will highly appreciate the following tip: IDL’s command line editing is broken beyond hope. But you can fix it, by running IDL under rlwrap, a wrapper for the excellent GNU readline library.
Download and install rlwrap from http://utopia.knoware.nl/~hlub/uck/rlwrap/ (on some systems you just need to run emerge rlwrap, or apt-get install rlwrap), and alias your idl command:
alias idl 'rlwrap -a -c idl'`` (csh)
alias idl='rlwrap -a -c idl'`` (bash)
From now on, you can
use long command lines that correctly wrap around;
type the first letters of a command and then PageUp to recall commands starting with these letters;
capitalize, uppercase or lowercase a word with Esc-C, Esc-U, Esc-L;
use command history across IDL sessions;
complete file names with Tab;
and use all other readline features that you are using in bash, octave, bc, gnuplot, ftp, etc.
2.5.17.4.2. Reading data into IDL arrays or structures
As an alternative to the method described above, there is also the possibility to read the data into structures. This makes some more operations possible, e.g., reading data from an IDL program where the command .r is not allowed.
IDL> pc_read_var_raw, obj=var, tags=tags
IDL> bb = pc_get_quantity ('B', var, tags)
IDL> jj = pc_get_quantity ('j', var, tags)
To read a snapshot VAR10 into the IDL structure ff, type:
IDL> pc_read_var, obj=ff, varfile='VAR10', /trimall
The option /trimall removes ghost zones from the data. You can see what data the structure contains by using the command tag_names:
IDL> print, tag_names(ff)
T X Y Z DX DY DZ UU LNRHO AA
One can access the individual variables by typing ff.varname, e.g.,
IDL> help, ff.aa
<Expression> FLOAT = Array[32, 32, 32, 3]
There are a number of files that read different data into structures. They are
placed in the directory $PENCIL_HOME/idl/files contains files to read different data into structures, e.g.,
- pc_read_var_raw, obj=var, tags=tags
Efficiently read a snapshot into an array.
- pc_read_var, obj=ff, /trimall
Read a snapshot into a structure.
- pc_read_ts, obj=ts
Read the time series into a structure.
- pc_read_xyaver, obj=xya ; pc_read_xzaver, obj=xza ; pc_read_yzaver, obj=yza
Read 1-D time series into a structure.
- pc_read_const, obj=cst
Read code constants into a structure.
- pc_read_pvar, obj=fp
Read particle data into a structure.
- pc_read_param, obj=par
Read startup parameters into a structure.
- pc_read_param, obj=par2, /param2
Read runtime parameters into a structure.
Other options are documented in the source code of the files.
For examples on how to use these routines, see GDL.
2.5.17.5. Python
The PENCIL CODE supports reading, processing and the visualization of data using python. A number of scripts are placed in the subfolder $PENCIL_HOME/python. A README file placed under that subfolder contains the information needed to read Pencil output data into python.
2.5.17.5.1. Installation
For modern operating systems, Python is generally installed together with the system. If not, it can be installed via your preferred package manager or downloaded from the website https://www.python.org/. For convenience, it is strongly recommend to also install IPython, which is a more convenient console for Python. You will also need the Python packages NumPy, matplotlib, h5py, Tk and often Dill.
Perhaps the easiest way to obtain all the required software mentioned above is to install either Continuum’s Anaconda or Enthought’s Canopy. These Python distributions also provide (or indeed are) integrated graphical development environments.
Another way of installing libraries, particularly on a cluster without root privileges, is to use pip or pip3: pip install h5py or pip3 install h5py.
In order for Python to find the Pencil Code commands, you will have to set the environment variable PYTHONPATH:
export PYTHONPATH=${PENCIL_HOME}/python
# or
export PYTHONPATH=${PENCIL_HOME}/python:${PYTHONPATH}
Normally, you will add one of these lines to your .bashrc file.
In addition, you have to import the pencil module each time you start a Python shell:
import pencil as pc
The next two paragraphs show how you can include this and some other imports into your Python interpreter setup, so they are automatically run when you start IPython or the Python REPL.
2.5.17.5.2. ipythonrc
When using IPython, some Pencil Code users add the following line to their .ipython/ipythonrc (create the file if it doesn’t exist):
import_all pencil
and in addition add to their .ipython/profile_default/startup/init.py the lines:
import pencil as pc
import numpy as np
import pylab as plt
import matplotlib
from matplotlib import rc
plt.ion()
matplotlib.rcParams['savefig.directory'] = ''
2.5.17.5.3. .pythonrc
If you don’t have IPython and cannot install it (e.g., on some cluster), you can instead edit your .pythonrc:
#!/usr/bin/python
import numpy as np
import pylab as plt
import pencil as pc
import atexit
#import readline
import rlcompleter
# enables search with Ctrl+r in the history
try:
import readline
except ImportError:
print "Module readline not available."
else:
import rlcompleter
readline.parse_and_bind("tab: complete")
# enables command history
historyPath = os.path.expanduser("~/.pyhistory")
def save_history(historyPath=historyPath):
import readline
readline.write_history_file(historyPath)
if os.path.exists(historyPath):
readline.read_history_file(historyPath)
atexit.register(save_history)
del os, atexit, readline, rlcompleter, save_history, historyPath
plt.ion()
and create the file .pythonhistory and add to your .bashrc:
export PYTHONSTARTUP=~/.pythonrc
However, none of this is required to use the Pencil Code Python modules.
2.5.17.5.4. Pencil Code Commands in General
For a list of all Pencil Code commands start IPython and type pc. <TAB> (as with auto completion). To access the help of any command just type the command followed by a ‘?’ (no spaces), e.g.,
In [1]: pc.math.dot?
Signature: pc.math.dot(a, b)
Docstring:
Take dot product of two pencil-code vectors a and b.
call signature:
dot(a, b):
Keyword arguments:
*a*, *b*:
Pencil-code vectors with shape [3, mz, my, mx].
File: ~/pencil-code/python/pencil/math/vector_multiplication.py
Type: function
You can also use help(pc.dot) for a more complete documentation of the command.
There are various reading routines for the Pencil Code data. All of them return an object with the data. To store the data into a user defined variable type, e.g.,
In [1]: ts = pc.read.ts()
Most commands take some arguments. For most of them there is a default value, e.g.,
In [1]: pc.read.ts(file_name='time_series.dat', datadir='data')
2.5.17.5.5. Reading and Plotting Time Series
Reading the time series file is very easy. Simply type
In [1]: ts = pc.read.ts()
and Python stores the data in the variable ts. The physical quantities are members of the object ts and can be accessed accordingly, e.g., ts.t, ts.emag. To check which other variables are stored simply do the tab auto completion ts. <TAB>.
Plot the data with the matplotlib commands:
In [1]: plt.plot(ts.t, ts.emag)
The standard plots are not perfect and need a little polishing. See the online wiki for a few examples on how to make pretty plots (https://github.com/pencil-code/pencil-code/wiki/PythonForPencil). You can save the plot into a file using the GUI or with
In [1]: plt.savefig('plot.eps')
2.5.17.5.6. Reading and Plotting VAR files and slice files
Read var files:
In [1]: var = pc.read.var()
Read slice files:
In [1]: slices = pc.read.slices(field='bb1', extension='xy')
This returns the object slices with indices slices[nTimes, my, mx] and the time array t.
If you want to plot, e.g., the x-component of the magnetic field at the central plane simply type:
In [1]: plt.imshow(var.bb[0, 128, :, :].T, origin='lower', extent=[-4, 4, -4, 4])
For a complete list of arguments of plt.imshow refer to its documentation.
For a more interactive plot use:
In [1]: pc.visu.animate_interactive(slices.xy.bb1, t)
Be aware
arrays from the reading routines are ordered f[nvar, mz, my, mx], i.e. reversed to IDL. This affects reading var files and slice files.
2.5.18. Running on multi-processor computers
The code is parallelized using MPI for a simple domain decomposition (data-parallelism), which is a straight-forward and very efficient way of parallelizing finite-difference codes. The current version has a few restrictions, which need to be kept in mind when using the MPI features.
The global number of grid points (but excluding the ghost zones) in a given direction must be an exact multiple of the number of processors you use in that direction. For example if you have nprocy=8 processors for the \(y\) direction, you can run a job with nygrid=64 points in that direction, but if you try to run a problem with nygrid=65 or nygrid=94, the code will complain about an inconsistency and stop. (So far, this has not been a serious restriction for us.)
2.5.18.1. How to run a sample problem in parallel
To run the sample problem in the directory samples/conv-slab on 16 CPUs, you need to do the following (in that directory):
Edit
src/Makefile.localand replaceMPICOMM = nompicomm \
by
MPICOMM = mpicomm \
Edit
src/cparam.localand replaceinteger, parameter :: ncpus=1, nprocy=1, nprocz=ncpus/nprocy, nprocx=1 integer, parameter :: nxgrid=32, nygrid=nxgrid, nzgrid=nxgrid \
by
integer, parameter :: ncpus=16, nprocy=4, nprocz=ncpus/nprocy, nprocx=1 integer, parameter :: nxgrid=128, nygrid=nxgrid, nzgrid=nxgrid \
The first line specifies a \(4{\times}4\) layout of the data in the \(y\) and \(z\) direction. The second line increases the resolution of the run because running a problem as small as \(32^3\) on 16 CPUs would be wasteful. Even \(128^3\) may still be quite small in that respect. For performance timings, one should try and keep the size of the problem per CPU the same, so for example \(256^3\) on 16 CPUs should be compared with \(128^3\) on 2 CPUs.
Recompile the code
unix> (cd src; make)
Run it
unix> start.csh unix> run.csh
Make sure that all CPUs see the same data/ directory; otherwise things will go wrong.
Remember that in order to visualize the full domain with IDL (rather than just the domain processed and written by one processor), you need to use rall.pro instead of r.pro.
2.5.18.2. Hierarchical networks (e.g., on Beowulf clusters)
On big Beowulf clusters, a group of nodes is often connected with a switch of modest speed, and all these groups are connected via a \(n\) times faster uplink switch. When bandwidth-limited, it is important to make sure that consecutive processors are mapped onto the mesh such that the load on the uplink is \(\lesssim n\) times larger than the load on the slower switch within each group. On a 512 node cluster, where groups of 24 processors are linked via fast ethernet switches, which in turn are connected via a Gigabit uplink (\(\sim 10\) times faster), we found that nprocy=4 is optimal. For 128 processors, for example we find that \(nprocy \times nprocz=4\times32\) is the optimal layout. For comparison, \(8\times16\) is 3 times slower, and \(16\times8\) is 17 (!) times slower. These results can be understood from the structure of the network, but the basic message is to watch out for such effects and to try varying nprocy and nprocz.
In cases where nygrid > nzgrid it may be advantageous to swap the ordering of processor numbers. This can be done by setting lprocz_slowest = F.
2.5.18.3. Extra workload caused by the ghost zones
Normally, the workload caused by the ghost zones is negligible. However, if one increases the number of processors, a significant fraction of work is done in the ghost zones. In other words, the effective mesh size becomes much larger than the actual mesh size.
Consider a mesh of size \(N_w=N_x\times N_y\times N_z\), and distribute the task over \(P_w=P_x\times P_y\times P_z\) processors. If no communication were required, the number of points per processor would be
However, for finite difference codes some communication is required, and the amount of communication depends on spatial order of the scheme, \(Q\). The PENCIL CODE works by default with sixth order finite differences, so \(Q=6\), i.e., one needs 6 ghost zones, 3 on each end of the mesh. With \(Q\neq0\) the number of points per processor is
There is efficient scaling only when
In the special case where \(N_x=N_y=N_z\equiv N=N_w^{1/3}\), with \(P_x=1\) and \(P_y=P_z\equiv P=P_w^{1/2}\), we have
For \(N=128\) and \(Q=6\) the effective mesh size exceeds the actual mesh size by a factor
These factors are listed in Table 2.3.
P N |
128 |
256 |
512 |
1024 |
2048 |
|---|---|---|---|---|---|
1 |
1.15 |
1.07 |
1.04 |
1.02 |
1.01 |
2 |
1.19 |
1.09 |
1.05 |
1.02 |
1.01 |
4 |
1.25 |
1.12 |
1.06 |
1.03 |
1.01 |
8 |
1.34 |
1.16 |
1.08 |
1.04 |
1.02 |
16 |
1.48 |
1.22 |
1.11 |
1.05 |
1.03 |
32 |
1.68 |
1.31 |
1.15 |
1.07 |
1.04 |
64 |
1.98 |
1.44 |
1.21 |
1.10 |
1.05 |
128 |
2.45 |
1.64 |
1.30 |
1.14 |
1.07 |
256 |
3.21 |
1.93 |
1.43 |
1.20 |
1.10 |
512 |
4.45 |
2.40 |
1.62 |
1.29 |
1.14 |
Ideally, one wants to keep the work in the ghost zones at a minimum. If one accepts that 20–25% of work are done in the ghost zones, one should use 4 processors for \(128^3\) mesh points, 16 processors for \(256^3\) mesh points, 64 processors for \(512^3\) mesh points, 256 processors for \(1024^3\) mesh points, and 512 processors for \(1536^3\) mesh points.
2.5.19. Running in double-precision
With many compilers, you can easily switch to double precision (8-byte floating point numbers) as follows.
Add the lines
# Use double precision
REAL_PRECISION = double
to src/Makefile.local and (re-)run pc_setupsrc.
If REAL_PRECISION is set to double, the flag FFLAGS_DOUBLE
is appended to the Fortran compile flags.
The default for FFLAGS_DOUBLE is -r8, which works for
g95 or ifort; for gfortran, you need to make sure that
FFLAGS_DOUBLE is set to -fdefault-real-8.
You can see the flags in src/Makefile.inc, which is the first place
to check if you have problems compiling for double precision.
medskip
Using double precision might be important in turbulence runs where the resolution is \(256^3\) meshpoints and above (although such runs often seem to work fine at single precision).
To continue working in double precision, you just say
lread_from_other_prec=T in run_pars; see
After-changing-REALPRECISION.
2.5.20. Power spectrum
Given a real variable \(u\), its Fourier transform \(\tilde{u}\) is given by
where
when \(L\) is the size of the simulation box. The three-dimensional power spectrum \(P(k)\) is defined as
where
Note that we can only reasonably calculate \(P(k)\) for \(k < \pi N_x/L_x\).
To get power spectra from the code, edit run.in and add for example
the following lines
dspec=5., ou_spec=T, ab_spec=T !(for energy spectra)
oned=T
under run_pars.
The kinetic (vel_spec) and magnetic (mag_spec) power spectra
will now be calculated every 5.0 (dspec) time units.
The Fourier spectra is calculated using fftpack.
In addition to calculating the three-dimensional
power spectra also the one-dimensional power spectra will be calculated
(oned).
In addition one must edit
src/Makefile.local and add the lines
FOURIER = fourier_fftpack
POWER = power_spectrum
Running the code will now create the files powerhel_mag.dat and
power_kin.dat containing the three-dimensional
magnetic and kinetic power spectra respectively. In addition to these
three-dimensional files we will also find the one-dimensional files
powerbx_x.dat,
powerby_x.dat,
powerbz_x.dat,
powerux_x.dat,
poweruy_x.dat and
poweruz_x.dat.
In these files the data are stored such that the first line contains the
time of the snapshot, the following nxgrid/2 numbers represent the
power at each wavenumber, from the smallest to the largest.
If several snapshots have been saved, they are being stored immediately
following the preceding snapshot.
You can read the results with the idl procure power, like this:
power,'_kin','_mag',k=k,spec1=spec1,spec2=spec2,i=n,tt=t,/noplot
power,'hel_kin','hel_mag',k=k,spec1=spec1h,spec2=spec2h,i=n,tt=t,/noplot
If powerhel is invoked, krms is written during the first computation.
The relevant output file is power_krms.dat.
This is needed for a correct calculation of \(k\) used in the realizability
conditions.
A caveat of the implementation of Fourier transforms in the Pencil Code is that, due to the parallelization, the permitted resolution is limited to the case when one direction is an integer multiple of the other. So, it can be done for
Nx = n*Ny
Unfortunately, for some applications one wants Nx < Ny. Wlad experimented
with arbitrary resolution by interpolating \(x\) to the same resolution of \(y\)
prior to transposing, then transform the interpolated array and then
interpolating it back (check fourier_transform_y in
fourier_fftpack.f90).
A feature of our current implementation with \(x\) parallelization is
that fft_xyz_parallel_3D requires nygrid to be an integer
multiple of nprocy*nprocz.
Examples of good mesh layouts are listed in Table 2.4.
ny |
nprocx |
nprocy |
nprocz |
ncpus |
256 |
1 |
16 |
16 |
256 |
256 |
2 |
16 |
16 |
512 |
256 |
4 |
16 |
16 |
1024 |
256 |
8 |
16 |
16 |
2048 |
288 |
2 |
16 |
18 |
576 |
512 |
2 |
16 |
32 |
1024 |
512 |
4 |
16 |
16 |
1024 |
512 |
4 |
16 |
32 |
2048 |
576 |
4 |
18 |
32 |
2304 |
576 |
8 |
18 |
32 |
4608 |
576 |
16 |
18 |
32 |
9216 |
1024 |
4 |
32 |
32 |
4096 |
1024 |
4 |
16 |
64 |
4096 |
1024 |
8 |
16 |
32 |
4096 |
1152 |
4 |
36 |
32 |
4608 |
1152 |
4 |
32 |
36 |
4608 |
2304 |
2 |
32 |
72 |
4608 |
2304 |
4 |
36 |
64 |
9216 |
2304 |
4 |
32 |
72 |
9216 |
To visualize with IDL just type
power and you get the last snapshot of the three-dimensional
power spectrum. See head of $PENCIL_HOME/idl/power.pro
for options to power.
By default, the spectra for the initial times time is not outputted,
but it can sometimes be useful to have it.
In that case, one can put lspec_start=T.
2.5.21. Other power spectra
Over the years, many more spectra have been outputted and not everything is immediately documented. Here some examples:
data/powerhel_mag.dat
data/powerhel_kin.dat
data/power_saffman_ub.dat
data/power_saffman_mag.dat
data/power_mag.dat
data/power_krms.dat
data/power_kin.dat
Here, the first two are helicity spectra that come for free
(in addition to those with _kin and _mag) when
one invokes ou_spec=T and ab_spec=T.
The ones with _saffman refer to the spectra related to Saffman-like
invariants such as the Hosking integral (which refers to power_mag.dat)
and the cross-helicity Saffman-like invariant (power_saffman_ub.dat).
Their slopes for \(k\to0\) are the invariants divided by \(2\pi^2\); see
the papers by Hosking & Schekochihin (2021) as well as Zhou et al. (2022).
2.5.22. Structure functions
We define the p-th order longitudinal structure function of \(\vec{u}\) as
while the transverse is
Edit run.in and add for example the following lines
dspec=2.3,
lsfu=T, lsfb=T, lsfz1=T, lsfz2=T
under run_pars.
The velocity (lsfu), magnetic (lsfb) and Elsasser
(lsfz1 and lsfz2) structure functions will
now be calculated every 2.3 (dspec) time unit.
In addition one must edit
src/Makefile.local and add the line
STRUCT_FUNC = struct_func
In src/cparam.local, define lb_nxgrid and make
sure that
nxgrid = nygrid = nzgrid = 2**lb_nxgrid
E.g.
integer, parameter :: lb_nxgrid=5
integer, parameter :: nxgrid=2**lb_nxgrid,nygrid=nxgrid,nzgrid=nxgrid
Running the code will now create the files:
sfu-1.dat,sfu-2.dat,sfu-3.dat(velocity),sfb-1.dat,sfb-2.dat,sfb-3.dat(magnetic field),sfz1-1.dat,sfz1-2.dat,sfz1-3.dat(first Elsasser variable),sfz2-1.dat,sfz2-2.dat,sfz2-3.dat(second Elsasser variable),
which contains the data of interest.
The first line in each file contains the time \(t\)
and the number qmax, such that the largest moment
calculated is qmax-1.
The next imax numbers represent the first moment
structure function for the first snapshot, here
The next imax numbers contain the second moment structure function,
and so on until qmax-1.
The following imax numbers then contain the data of the
signed third order structure function i.e.
\(S^3_{\rm long}(l)=\left< [u_x(x{+}l,y,z)-u_x(x,y,z)]^3 \right>\).
The following imax × qmax × 2 numbers are zero if
nr_directions=1
(default), otherwise they are the same data as above but for the
structure functions calculated in the y and z directions.
If the code has been run long enough as to calculate several snapshots, these snapshots will now follow, being stored in the same way as the first snapshot.
To visualize with IDL just type
structure and you get the time-average of the first order longitudinal
structure function (be sure that
pencil-runs/forced/idl/ is in IDL_PATH).
See head of pencil-runs/forced/idl/structure.pro
for options to structure.
2.5.23. Particles
The PENCIL CODE has modules for tracer particles and for dust particles (see Sect.
Particles). The particle modules are chosen by setting the
value of the variable PARTICLES in Makefile.local to either
particles_dust or particles_tracers. For the former case
each particle has six degrees of freedom, three positions and three velocities.
For the latter it suffices to have only three position variables as the
velocity of the particles are equal to the instantaneous fluid velocity at
that point. In addition one can choose to have several additional internal
degrees of freedoms for the particles. For example one can temporally evolve
the particles radius by setting PARTICLES_RADIUS to
particles_radius in Makefile.local.
All particle infrastructure is controlled and organized by the
Particles_main module. This module is automatically selected by
Makefile.src if PARTICLES is different from
noparticles. Particle modules are compiled as a separate library.
This way the main part of the PENCIL CODE only needs to know about the
particles_main.a library, but not of the individual particle modules.
For a simulation with particles one must in addition define a few parameters
in cparam.local. Here is a sample of cparam.local for a
parallel run with 2,000,000 particles:
integer, parameter :: ncpus=16, nprocy=4, nprocz=4, nprocx=1
integer, parameter :: nxgrid=128, nygrid=256, nzgrid=128
integer, parameter :: npar=2000000, mpar_loc=400000, npar_mig=1000
integer, parameter :: npar_species=2
The parameter npar is the number of particles in the simulation,
mpar_loc is the number of particles that is allowed on each
processor and npar_mig is the number of particles that are allowed
to migrate from one processor to another in any time-step. For a
non-parallel run it is enough to specify npar. The number of
particle species is set through npar_species (assumed to be one if
not set). The particle input parameters are given in start.in and
run.in. Here is a sample of the particle part of start.in for
dust particles:
/
&particles_init_pars
initxxp='gaussian-z', initvvp='random'
zp0=0.02, delta_vp0=0.01, eps_dtog=0.01, tausp=0.1
lparticlemesh_tsc=T
/
The initial positions and velocities of the dust particles are set in initxxp and initvvp. The next four input parameters are further specifications of the initial condition. Interaction between the particles and the mesh, e.g., through drag force or self-gravity, require a mapping of the particles on the mesh. The PENCIL CODE currently supports NGP (Nearest Grid Point, default), CIC (Cloud in Cell, set lparticlemesh_cic=T) and TSC (Triangular Shaped Cloud, set lparticlemesh_tsc=T). See Youdin & Johansen (2007) for details.
Here is a sample of the particle part of run.in (also for dust
particles):
/
&particles_run_pars
ldragforce_gas_par=T
cdtp=0.2
/
The logical ldragforce_gas_par determines whether the dust particles influence the gas with a drag force. cdtp tells the code how many friction times should be resolved in a minimum time-step.
The sample run samples/sedimentation/ contains the latest setup for
dust particles.
2.5.23.1. Particles in parallel
The particle variables (e.g., \(\vec{x}_i\) and \(\vec{v}_i\)) are
kept in the arrays fp and dfp. For parallel runs,
particles must be able to move from processor to processor as they pass out
of the \((x,y,z)\)-interval of the local processor. Since not all
particles are present at the same processor at the same time (hopefully),
there is some memory optimization in making fp not big enough to
contain all the particles at once. This is achieved by setting the code
variable mpar_loc less than npar in cparam.local
for parallel runs. When running with millions of particles, this trick is
necessary to keep the memory need of the code down.
The communication of migrating particles between the processors happens as
follows (see the subroutine redist_particles_procs in
particles_sub.f90):
In the beginning of each time-step all processors check if any of their particles have crossed the local \((x,y,z)\)-interval. These particles are called migrating particles. A run can have a maximum of npar_mig migrating particles in each time-step. The value of npar_mig must be set in
cparam.local. The number should (of course) be slightly larger than the maximum number of migrating particles at any time-step during the run. The diagnostic variable nmigmax can be used to output the maximum number of migrating particles at a given time-step. One can set lmigration_redo=T in &particles_run_pars to force the code to redo the migration step if more than npar_mig want to migrate. This does slow the code down somewhat, but has the benefit that the code does not stop when more than npar_mig particles want to migrate.The index number of the receiving processor is then calculated. This requires some assumption about the grid on other processors and will currently not work for nonequidistant grids. Particles do not always pass to neighboring processors as the global boundary conditions may send them to the other side of the global domains (periodic or shear periodic boundary conditions).
The migrating particle information is copied to the end of fp, and the empty spot left behind is filled up with the particle of the highest index number currently present at the processor.
Once the number of migrating particles is known, this information is shared with neighboring processors (including neighbors over periodic boundaries) so that they all know how many particles they have to receive and from which processors.
The communication happens as directed MPI communication. That means that processors 0 and 1 can share migrating particles at the same time as processors 2 and 3 do it. The communication happens from a chunk at the end of fp (migrating particles) to a chunk that is present just after the particle of the highest index number that is currently at the receiving processor. Thus the particles are put directly at their final destination, and the migrating particle information at the source processor is simply overwritten by other migrating particles at the next time-step.
Each processor keeps track of the number of particles that it is responsible for. This number is stored in the variable npar_loc. It must never be larger than mpar_loc (see above). When a particle leaves a processor, npar_loc is reduced by one, and then increased by one at the processor that receives that particle. The maximum number of particles at any processor is stored in the diagnostic variable nparmax. If this value is not close to npar/ncpus, the particles have piled up in such a way that computations are not evenly shared between the processors. One can then try to change the parallelization architecture (nprocy and nprocz) to avoid this problem.
In simulations with many particles (comparable to or more than the number of grid cells), it is crucial that particles are shared relatively evenly among the processors. One can as a first approach attempt to not parallelize directions with strong particle density variations. However, this is often not enough, especially if particles clump locally.
Alternatively one can use Particle Block Domain Decomposition (PBDD, see Johansen et al. 2011). The steps in Particle Block Domain Decomposition scheme are as follows:
The fixed mesh points are domain-decomposed in the usual way (with ncpus = nprocx * nprocy * nprocz).
Particles on each processor are counted in bricks of size nbx * nby * nbz (typically nbx = nby = nbz = 4).
Bricks are distributed among the processors so that each processor has approximately the same number of particles.
Adopted bricks are referred to as blocks.
The PENCIL CODE uses a third order Runge-Kutta time-stepping scheme. In the beginning of each sub-time-step particles are counted in blocks and the block counts communicated to the bricks on the parent processors. The particle density assigned to ghost cells is folded across the grid, and the final particle density (defined on the bricks) is communicated back to the adopted blocks. This step is necessary because the drag force time-step depends on the particle density, and each particle assigns density not just to the nearest grid point, but also to the neighboring grid points.
In the beginning of each sub-time-step the gas density and gas velocity field is communicated from the main grid to the adopted particle blocks.
Drag forces are added to particles and back to the gas grid points in the adopted blocks. This partition aims at load balancing the calculation of drag forces.
At the end of each sub-time-step the drag force contribution to the gas velocity field is communicated from the adopted blocks back to the main grid.
Particle Block Domain Decomposition is activated by setting
PARTICLES = particles_dust_blocks and
PARTICLES_MAP = particles_map_blocks in Makefile.local.
A sample of cparam.local for Particle Block Domain Decomposition can
be found in samples/sedimentation/blocks:
integer, parameter :: ncpus=4, nprocx=2, nprocy=2, nprocz=1
integer, parameter :: nxgrid=32, nygrid=32, nzgrid=32
integer, parameter :: npar=10000, mpar_loc=5000, npar_mig=100
integer, parameter :: npar_species=4
integer, parameter :: nbrickx=4, nbricky=4, nbrickz=4, nblockmax=32
The last line defines the number of bricks in the total domain – here we divide the grid into 4 x 4 x 4 bricks each of size 8 x 8 x 8 grid points. The parameter nblockmax tells the code the maximum number of blocks any processor may adopt. This should not be so low that there is not room for all the bricks with particles, nor so high that the code runs out of memory.
2.5.23.2. Large number of particles
When dealing with large number of particles, one needs to make sure that the number of particles npar is less than the maximum integer that the compiler can handle with. The maximum integer can be checked by the Fortran intrinsic function huge:
program huge_integers
print *, huge(0_4) ! for default Fortran integer (32 Bit)
print *, huge(0_8) ! for 64 Bit integer in Fortran
end program huge_integers
If the number of particles npar is larger than default maximum integer, one can promote the maximum integer to 64 Bit by setting:
integer(kind=8), parameter :: npar=4294967296
in the cparam.local file. This works because the data type of
npar is only set here. It is worth noting that one should not use
the flag:
FFLAGS += -integer-size 64
to promote all the integers to 64 Bit. This will break the Fortran-C interface. One should also make sure that npar_mig <= npar/ncpus. It is also beneficial to set mpar_loc = 2*npar/ncpus.
Sometimes one may encounter the following error, “additional relocation overflows omitted from the output” due to the 2G memory limit (caused by large static array). It is mpar_loc that determines the usage of memory instead of npar. There are two ways to resolve this issue:
- Use a specific memory model to generate code and store data by setting the
following for Intel compiler in your configuration file:
FFLAGS += -shared-intel -mcmodel=medium
This will, however, affect the performance of the code. This method can handle at least the following setup:
integer, parameter :: ncpus=256, nprocx=4, nprocy=8, nprocz=ncpus/(nprocx*nprocy) integer, parameter :: nxgrid=1024, nygrid=nxgrid, nzgrid=nxgrid integer(kind=ikind8), parameter :: npar=124999680 integer, parameter :: npar_stalk=100000, npar_mig=npar/10 integer, parameter :: mpar_loc=npar/5
- Increase ncpu and decrease mpar_loc. For
npar=124999680, ncpu=1024 is needed.
integer, parameter :: ncpus=1024, nprocx=8, nprocy=8, nprocz=ncpus/(nprocx*nprocy) integer, parameter :: nxgrid=1024, nygrid=nxgrid, nzgrid=nxgrid integer(kind=ikind8), parameter :: npar=124999680 integer, parameter :: mpar_loc=5*npar/ncpus integer, parameter :: npar_stalk=100000, npar_mig=npar/ncpus
It is worth noting that even without particles, a simulation with 1024^3 resolution requires at least 512 CPUs to be compiled.
2.5.23.3. Random number generator
There are several methods to generate random number in the code. It is worth
noting that when simulating coagulation with the super-particle approach, one
should use the intrinsic random number generator of FORTRAN instead of the one
implemented in the code. When invoking random_number_wrapper, there
will be back-reaction to the gas flow. This unexpected back-reaction can be
tracked by inspecting the power spectra, which exhibits the oscillation at
the tail. To avoid this, one should set
luser_random_number_wrapper=F under the module
particles_coag_run_pars in run.in.
2.5.24. Non-cartesian coordinate systems
Spherical coordinates are invoked by adding the following line in the file
start.in:
&init_pars
coord_system='spherical_coords'
One can also invoke cylindrical coordinates by saying cylindrical_coords instead. In practice, the names \((x,y,z)\) are still used, but they then refer to \((r,\theta,\phi)\) or \((r,\phi,z)\) instead.
When working with curvilinear coordinates it is convenient to use differential operators in the non-coordinate basis, so the derivatives are taken with respect to length, and not in a mixed fashion with respect to length for \(\partial/\partial r\) and with respect to angle for \(\partial/\partial \theta\) and \(\partial/\partial \phi\). The components in the non-coordinate basis are denoted by hats, see, e.g., [MTW], p. 213; see also Appendix B of [MTBM09].
For spherical polar coordinates the only nonvanishing Christoffel symbols (or connection coefficients) are:
The Christoffel symbols enter as correction terms for the various differential operators in addition to a term calculated straightforwardly in the non-coordinate basis. The derivatives of some relevant Christoffel symbols are:
Further details are given in Curvilinear-coordinates.
2.6. The equations
The equations solved by the PENCIL CODE are basically the standard
compressible MHD equations. However, the modular structure allows
some variations of the MHD equations, as well as switching off
some of the equations or individual terms of the equation (nomagnetic,
noentropy, etc.).
In this section the equations are presented in their most complete form. It may be expected that the code can evolve most subsets or simplifications of these equations.
2.6.1. Continuity equation
In the code the continuity equation, \(\partial\rho/\partial t + \nabla\cdot(\rho\mathbf{u}) = 0\), is written in terms of \(\ln\rho\),
Here \(\rho\) denotes density, \(\uv\) the fluid velocity, \(t\) is time and \(\De/\De t \equiv \partial/\partial t + \mathbf{u}\cdot\nabla\) is the advective derivative.
2.6.2. Equation of motion
In the equation of motion, using a perfect gas, the pressure term, can be expressed as \(-\rho^{-1}\grad p = -\cs^2(\grad s/c_p+\grad\ln\rho)\), where the squared sound speed is given by
and \(\gamma=c_p/c_v\) is the ratio of specific heats, or adiabatic index. Note that \(\cs^2\) is proportional to the temperature with \(\cs^2=(\gamma-1)c_p T\).
The equation of motion is accordingly
Here \(\Phi_{\rm grav}\) is the gravity potential, \(\jv\) the electric current density, \(\Bv\) the magnetic flux density, \(\nu\) is kinematic viscosity, \(\zeta\) describes a bulk viscosity, and, in Cartesian coordinates
is the rate-of-shear tensor that is traceless, because it can be written as the generic rate-of-strain tensor minus its trace. In curvilinear coordinates, we have to replace partial differentiation by covariant differentiation (indicated by semicolons), so we write \({\mathsf S}_{ij}=\frac{1}{2}(u_{i;j}+u_{j;i})-\frac{1}{3}\delta_{ij}\Div\uv\).
The interpretation of the two viscosity terms varies greatly depending upon the Viscosity module used, and indeed on the parameters given to the module. See Bulkviscosity.
For isothermal hydrodynamics, see Entropy equation below.
2.6.3. Induction equation
Here \(\Av\) is the magnetic vector potential, \(\Bv\) = \(\boldsymbol{\nabla}\times\) \(\Av\) the magnetic flux density, \(\eta = 1/(\mu_0\sigma)\) is the magnetic diffusivity (\(\sigma\) denoting the electrical conductivity), and \(\mu_0\) the magnetic vacuum permeability. This form of the induction equation corresponds to the Weyl gauge \(\Phi=0\), where \(\Phi\) denotes the scalar potential.
2.6.4. Entropy equation
The current thermodynamics module entropy formulates the thermal part of the physics in terms of entropy \(s\), rather than thermal energy \(e\), which you may be more familiar with. Thus the two fundamental thermodynamical variables are \(\ln\rho\) and \(s\). The reason for this choice of variables is that entropy is the natural physical variable for (at least) convection processes: the sign of the entropy gradient determines convective (in)stability, the Rayleigh number is proportional to the entropy gradient of the associated hydrostatic reference solution, etc. The equation solved is
Here, \(T\) is temperature, \(c_p\) the specific heat at constant pressure, \(\Heat\) and \(\Cool\) are explicit heating and cooling terms, \(K\) is the radiative (thermal) conductivity, \(\zeta\) describes a bulk viscosity, and \(\Strain\) is the rate-of-shear tensor that is traceless.
In the entropy module we solve for the specific entropy, \(s\). The heat conduction term on the right hand side can be written in the form
where \(\chi = K/(\rho c_p)\) is the thermal diffusivity. The latter equation shows that the diffusivity for \(s\) is \(\gamma\chi\), which is what we have used in (2.3).
In an alternative formulation for a constant \(K\), the heat conduction term on the right hand side can also be written in the form
which is the form used when constant \(K\) is chosen.
Note that by setting \(\gamma=1\) and initially \(s=0\), one obtains an isothermal equation of state (albeit at some unnecessary expense of memory). Similarly, by switching off the evolution terms of entropy, one immediately gets polytropic behavior (if \(s\) was initially constant) or generalized polytropic behavior (where \(s\) is not uniform, but \(\partial s/\partial t = 0\)).
A better way to achieve isothermality is to use the noentropy
module.
2.6.4.1. Viscous heating
We can write the viscosity as the divergence of a tensor \(\tau_{ij,j}\),
where \(\tau_{ij}=2\nu\rho{\sf S}_{ij}\) is the stress tensor. The viscous power density \(P\) is
The term under the divergence is the viscous energy flux and the other term is the kinetic energy loss due to heating. The heating term \(+u_{i,j}\tau_{ij}\) is positive definite, because \(\tau_{ij}\) is a symmetric tensor and the term only gives a contribution from the symmetric part of \(u_{i,j}\), which is \(\frac{1}{2}(u_{i,j}+u_{j,i})\), so
But, because \({\sf S}_{ij}\) is traceless, we can add anything proportional to \(\delta_{ij}\) and, in particular, we can write
which is positive definite.
2.6.4.2. Alternative description
By setting pretend_lnTT=T in init_pars or
run_pars (i.e. the general part of the name list)
the logarithmic temperature is used instead of the entropy.
This has computational advantages when heat conduction
(proportional to \(K\grad T\)) is important.
Another alternative is to use another module, i.e. set
ENTROPY=temperature_idealgas in Makefile.local.
When pretend_lnTT=T is set, the entropy equation
is replaced by
where \(\mathbf{RHS}\) is the right hand side of equation (2.10).
2.6.5. Transport equation for a passive scalar
In conservative form, the equation for a passive scalar is
Here c denotes the concentration (per unit mass) of the passive scalar and cal D its diffusion constant (assumed constant). In the code this equation is solved in terms of \(\ln c\),
Using \(\ln c\) instead of c has the advantage that it enforces \(c>0\) for all times. However, the disadvantage is that one cannot have \(c=0\). For this reason we ended up using the non-logarithmic version by invoking PSCALAR=pscalar_nolog.
2.6.6. Bulk viscosity
For a monatomic gas it can be shown that the bulk viscosity vanishes. We therefore don’t use it in most of our runs. However, for supersonic flows, or even otherwise, one might want to add a shock viscosity which, in its simplest formulation, take the form of a bulk viscosity.
2.6.6.1. Shock viscosity
Shock viscosity, as it is used here and also in the Stagger Code of {AA}ke Nordlund, is proportional to positive flow convergence, maximum over five zones, and smoothed to second order,
where c_{rm shock} is a constant defining the strength of the shock viscosity. In the code this dimensionless coefficient is called nu_shock, and it is usually chosen to be around unity. Assume that the shock viscosity only enters as a bulk viscosity, so the whole stress tensor is then
Assume \(\nu=\mbox{const}\), but \(\zeta\neq\mbox{const}\), so
and
In the special case with periodic boundary conditions, we have \(2\langle\Strain^2\rangle=\langle\omv^2\rangle+{4\over3}\langle(\nabla\cdot\uv)^2\rangle\).
2.6.7. Equation of state
In its present configuration only hydrogen ionization is explicitly included. Other constituents (currently He and H:math:_2) can have fixed values. The pressure is proportional to the total number of particles, i.e.
It is convenient to normalize to the total number of H both in atomic and in molecular hydrogen, \(n_{\rm Htot}\equiv n_{\rm H}+2n_{\rm H_2}\), where \(n_{\rm HI}+n_{\rm HII}=n_{\rm H}\), and define \(x_{\rm e}\equiv n_{\rm e}/n_{\rm Htot}\), \(x_{\rm He}\equiv n_{\rm He}/n_{\rm Htot}\), and \(x_{\rm H_2}\equiv n_{\rm H_2}/n_{\rm Htot}\). Substituting \(n_{\rm H}=n_{\rm Htot}-2n_{\rm H_2}\), we have
This can be written in the more familiar form
where \({\cal R}=k_{\rm B}/m_{\rm u}\) and \(m_{\rm u}\) is the atomic mass unit (which is for all practical purposes the same as \(m_{\rm Htot}\)) and
is the mean molecular weight (which is here dimensionless; see Kippenhahn & Weigert 1990, p.102). The factor 4 is really to be substituted for 3.97153. Some of the familiar relations take still the usual form, in particular \(e=c_vT\) and \(h=c_pT\) with \(c_v={3\over2}{\cal R}/\mu\) and \(c_p={5\over2}{\cal R}/\mu\).
The number ratio, \(x_{\rm He}\), is more commonly expressed as the mass ratio, \(Y=m_{\rm He}n_{\rm He}/(m_{\rm H}n_{\rm Htot}+m_{\rm He}n_{\rm He})\), or \(Y=4x_{\rm He}/(1+4x_{\rm He})\), or \(4x_{\rm He}=(1/Y-1)^{-1}\). For example, \(Y=0.27\) corresponds to \(x_{\rm He}=0.092\) and \(Y=0.25\) to \(x_{\rm He}=0.083\). Note also that for 100% H$_2$ abundance, \(x_{\rm H_2}=1/2\).
In the following, the ionization fraction is given as \(y=n_{\rm e}/n_{\rm H}\), which can be different from \(x_{\rm e}\) if there is H$_2$. Substituting for \(n_{\rm H}\) in terms of \(n_{\rm Htot}\) yields \(y=x_{\rm e}/(1-2x_{\rm H_2})\).
2.6.8. Ionization
This part of the code can be invoked by setting EOS=eos_ionization
(or EOS=eos_temperature_ionization) in the Makefile.local file.
The equation of state described below works for variable ionization, and the
entropy offset is different from that used in (2.6), which
is now no longer valid. As a replacement, one can use
EOS=eos_fixed_ionization, where the degree of ionization can be given
by hand. Here the normalization of the entropy is the same as for
EOS=eos_ionization. This case is described in more detail
below. [11]
We treat the gas as being composed of partially ionized hydrogen and neutral helium. These are four different particle species, each of which regarded as a perfect gas.
The ionization fraction \(y\), which gives the ratio of ionized hydrogen to the total amount of hydrogen \(n_{\rm H}\), is obtained from the Saha equation which, in this case, may be written as
The temperature T cannot be obtained directly from the Pencil Code’s independent variables \(\ln\rho\) and s, but is itself dependent on \(y\). Hence, the calculation of \(y\) essentially becomes a root finding problem.
The entropy of a perfect gas consisting of particles of type \(i\) is known from the Sackur-Tetrode equation
Here \(N_i\) is the number of particles of a single species and \(n_{\rm tot}\) is the total number density of all particle species.
In addition to the individual entropies we also have to take the entropy of mixing, \(S_{\rm mix}=-N_{\rm tot}k_{\rm B}\sum_ip_i\ln p_i\), into account. Summing up everything, we can get a closed expression for the specific entropy s in terms of \(y\), \(\ln\rho\) and \(T\), which may be solved for T.
Fig. 2.4 Dependence of temperature on entropy for different values of the density.
For given \(\ln\rho\) and s we are then able to calculate the ionization fraction \(y\) by finding the root of
In the ionized case, several thermodynamic quantities of the gas become dependent on the ionization fraction \(y\) such as its pressure, \(P\!=(1\!+y+x_{\rm He})n_{\rm H}k_{\rm B}T\), and its internal energy, \(E=\frac{3}{2}(1+y+x_{\rm He})n_{\rm H}k_{\rm B}T+y\chi_{\rm H}\), where \(x_{\rm He}\) gives the ratio of neutral helium to the total amount of hydrogen. The dependence of temperature on entropy is shown in Fig. 2.4 for different values of the density.
For further details regarding the procedure of solving for the entropy
see S-Ioni in the appendix.
2.6.9. Ambipolar diffusion
Another way of dealing with ionization in the PENCIL CODE is through use of the neutrals module. That module solves the coupled equations of neutral and ionized gas, in a two-fluid model
where the subscripts \(n\) and \(i\) are for neutral and ionized, respectively. The terms \({\mathcal G}\) and \({\mathcal F}\), through which the two fluids exchange mass and momentum, are given by
In the above equations, \(\zeta\) is the ionization coefficient, \(\alpha\) is the recombination coefficient, and \(\gamma\) the collisional drag strength. By the time of writing (spring 2009), these three quantities are supposed constant. The electron pressure \(p_e\) is also assumed equal to the ion pressure. Only isothermal neutrals are supported so far.
In the code, (2.13) and (2.15) are solved in density.f90
and hydro.f90 respectively. (2.14) is solved in
neutraldensity.f90 and (2.16) in neutralvelocity.f90. The sample
1d-test/ambipolar-diffusion has the current setup for a two-fluid simulation with
ions and neutrals.
2.6.10. Combustion
The easiest entry into the subject of simulating combustion is through
samples/0d-tests/chemistry_H2_ignition_rkf
or samples/1d-tests/H2_flamespeed.
The former case is studying H2 ignition delay, while the second one
is focusing on H2 flame-speed.
If you want to study turbulent premixed flames, the recommended cases are
samples/2d-tests/2d_methane_flame and samples/turbulent_flame.
Here, the first of these examples is not really realistic since its only 2D,
but this does of course make it faster to try out.
Also, this case is easier to set up. The most realistic test case is
samples/turbulent_flame, which studies a full 3D hydrogen flame.
This test case requires that a set of smaller pre-runs are finalized
before the main simulation can be run (see the associated README file
for more details).
The chemistry_H2_ignition_rkf directory, for example, has the file
tran.dat, which contains the parameters characterizing the transport
properties, and chem.inp, which contains the NASA polynomials;
see Eq. 18 of [BHB11-18].
2.6.11. Radiative transfer
Here we only state the basic equations.
A full description about the implementation is given in
S-radi-trans and in the original paper by
Heinemann et al. (2006).
The basic equation for radiative transfer is
where
is the optical depth (\(s\) is the geometrical coordinate along the ray).
Note that radiative transfer is called also in start.csh,
and again each time a snapshot is being written, provided the output
of auxiliary variables is being requested lwrite_aux=T.
(Also, of course, the pencil check runs radiative transfer 7 times,
unless you put pencil_check_small=F.)
2.6.12. Self-gravity
The PENCIL CODE can consider the self-gravity of the fluid in the simulation box by adding the term
to the equation of motion. The self-potential \(\phi_{\rm self}\) (or just \(\phi\) for simplicity) satisfies Poisson’s equation
The solution for a single Fourier component at scale \(\vec{k}\) is
Here we have assumed periodic boundary conditions. The potential is obtained by Fourier-transforming the density, then finding the corresponding potential at that scale, and finally Fourier-transforming back to real space.
The \(x\)-direction in the shearing sheet is not strictly periodic, but is rather shear periodic with two connected points at the inner and outer boundary separated by the distance \(\Delta y(t)={\rm mod}[(3/2) \varOmega_0 L_x t,L_y]\) in the \(y\)-direction. We follow here the method from [Gammie2001] to allow for shear-periodic boundaries in the Fourier method for self-gravity. First we take the Fourier transform along the periodic \(y\)-direction. We then shift the entire \(y\)-direction by the amount \(\delta y(x)=\Delta y(t) x/L_x\) to make the \(x\)-direction periodic. Then we proceed with Fourier transforms along \(x\) and then \(z\). After solving the Poisson equation in Fourier space, we transform back to real space in the opposite order. We differ here from the method by [Gammie2001] in that we shift in Fourier space rather than in real space [12] . The Fourier interpolation formula has the advantage over polynomial interpolation in that it is continuous and smooth in all its derivatives.
2.6.13. Incompressible and anelastic equations
This part has not yet been documented and is still under development.
2.6.14. Dust equations
The code treats gas and dust as two separate fluids [13] . The dust and the gas interact through a drag force. This force can most generally be written as an additional term to the equation of motion as
where \(\tau_{\rm s}\) is the so-called stopping time of the considered dust species. This measures the coupling strength between dust and gas. In the Epstein drag regime
where \(a_{\rm d}\) is the radius of the dust grain and \(\rho_{\rm s}\) is the solid density of the dust grain.
Two other important effects work on the dust. The first is coagulation controlled by the discrete coagulation equation
In the code \(N\) discrete dust species are considered. Also the bins are logarithmically spaced in order to give better mass resolution. It is also possible to keep track of both number density and mass density of each bin, corresponding to having a variable grain mass in each bin.
Dust condensation is controlled by the equation
where \(N\) is the number of monomers in the dust grain (such as water molecules) and \(d\) is the physical dimension of the dust grain. The condensation time \(\tau_{\rm cond}\) is calculated from
where \(A_1\) is the surface area of a monomer, \(\alpha\) is the condensation efficiency, \(n_{\rm mon}\) is the number density of monomers in the gas, and \(S_{\rm mon}\) is the saturation level of the monomer given by
Here \(P_{\rm sat}\) is the saturated vapor pressure of the monomer. Currently only water ice has been implemented in the code.
All dust species fulfill the continuity equation
2.6.15. Cosmic ray pressure in diffusion approximation
Cosmic rays are treated in the diffusion approximation. The equation of state is \(p_{\rm c}=(\gamma_{\rm c})e_{\rm c}\) where the value of \(\gamma_{\rm c}\) is usually somewhere between \(14/9\) and \(4/3\). In the momentum equation (2.7) the cosmic ray pressure force, \(-\rho^{-1}\nabla p_{\rm c}\) is added on the right hand side, and \(e_{\rm c}\) satisfies the evolution equation
where \(Q_{\rm c}\) is a source term and
is an anisotropic diffusivity tensor.
In the non-conservative formulation of this code it is advantageous to expand the diffusion term using the product rule, i.e.,
where \(U_{{\rm c}\,i}=-\partial K_{ij}/\partial x_j\) acts like an extra velocity trying to straighten magnetic field lines. We can write this term also as \(\Uv_{\rm c}=-(K_\parallel-K_\perp)\nabla\cdot(\BBhat\BBhat)\), where the last term is a divergence of the dyadic product of unit vectors [14] . However, near magnetic nulls, this term can becomes infinite. In order to avoid this problem we are forced to limit \(\nabla\cdot(\BBhat\BBhat)\) , and hence \(|\Uv_{\rm c}|\), to the maximum possible value that can be resolved at a given resolution
A physically appealing way of limiting the maximum propagation speed is to restore an explicit time dependence in the equation for the cosmic ray flux, and to replace the diffusion term in (2.18) by a divergence of a flux that in turn obeys the equation
where \(K_{ij}=\tau\tilde{K}_{ij}\) would be the original diffusion tensor of (2.19), if the time derivative were negligible. Further details are described in Snodin et al. (2006).
2.6.16. Chiral MHD
At high energies, \(k_{\rm B} T \gtrsim 10~\mathrm{MeV}\), a quantum effect called the chiral magnetic effect (CME) can modify the MHD equations. The CME occurs in magnetized relativistic plasmas, in which the number density of left-handed fermions, \(n_{_{\rm L}}\), differs from the one of right-handed fermions, \(n_{_{\rm R}}\) (see e.g., Kharzeev et al. 2013 for a review). This asymmetry is described by the chiral chemical potential
where \(T\) is the temperature, \(k_{\rm B}\) is the Boltzmann constant, \(c\) is the speed of light, and \(\hbar\) is the reduced Planck constant. In the presence of a magnetic field, \(\mu_5\) leads to the occurrence of the current
where \(\alpha_\mathrm{em} \approx 1/137\) is the fine structure constant.
The chiral current (2.21) adds to the classical Ohmic current, leading to a modification of the Maxwell equations. As a result, the induction equation is extended by one additional term:
where the chiral chemical potential \(\mu_5\) is normalized such that \(\mu = (4 \alpha_\mathrm{em} /\hbar c) \mu_5\). The latter is determined by the evolution equation
where \(D_5\) is a chiral diffusion coefficient, \(\lambda\) the chiral feedback parameter, and \(\Gamma_{\rm\!f}\) the rate of chiral flipping reactions. All remaining evolution equations are the same as in classical MHD. Details on the derivation of the chiral MHD equations can be found in Boyarsky et al. (2015) and Rogachevskii et al. (2017).
In the Pencil Code, the chiral MHD equations can be solved by adding the line:
SPECIAL = special/chiral_mhd
to src/Makefile.local.
Further, for running the chiral MHD module, one needs to add:
&special_init_pars
initspecial='const', mu5_const=10.
to start.in and:
&special_run_pars
diffmu5=1e-4, lambda5=1e3, cdtchiral=1.0
to run.in, where exemplary values for the chiral parameters are chosen.
Caution should be taken when solving the chiral MHD equations numerically, since the evolution of the plasma can be strongly affected by the CME. In particular, the initial value of \(\mu\) is related to a small-scale dynamo instability. In order to resolve this instability in a domain of size \((2\pi)^3\), the minimum number of grid points is given as Mesh Reynolds number:
Also, the value of \(\lambda\) should not be chosen too small, since it scales inversely with the saturation magnetic helicity produced by a chiral dynamo. Hence, for a very small \(\lambda\) parameter, the Alfv'en time step becomes extremely small in the non-linear stage, which can lead to a code crash. More details on the numerical modeling of chiral MHD can be found in Schober et al., 2018.
2.6.17. Electromagnetism with displacement current
In electromagnetism, the displacement current is not neglected, and so we solve the equation
in addition to the induction equation \(\partial\Bv/\partial t=-\nabla\times\Ev\),
or its uncurled version \(\partial\Av/\partial t=-\Ev\).
Solving this equation is invoked by putting SPECIAL=special/disp_current.
In the code, there is the line df(l1:l2,m,n,iex:iez)=df(l1:l2,m,n,iex:iez)+c_light2*(p%curlb-mu0*p%jj_ohm) where c_light2 is the speed of light squared as input parameter, so
it is usually not computed self-consistently from the actual speed of light,
which would be known once we use physical dimensions.
Furthermore, p%curlb is the curl of \(\Bv\) (which is now not
the electric current), while p%jj_ohm is the Ohmic current, obtained
by solving \(\Jv = \sigma(\Ev + \uv \times \Bv)\), and \(\sigma\) is the conductivity
(which can be time-dependent).
The vacuum permeability is mu0.
The electric field obtained in this way, which we now call the pseudo-electric field, \(\tilde{\Ev}\), does in general not obey the equation for the charge density, \(\rho_{\rm e}\),
The actual electric field \(\Ev\) can be obtained from the pseudo-electric field \(\tilde{\Ev}\) as
where \(\phi\) is obtained by solving a Poisson-like equation
where \(\rho_{\rm e}\) is obtained through time-integration of the charge continuity equation
which, in turn, follows from Eq. (2.28) by taking its divergence.
2.6.18. Particles
Particles are entities that each have a space coordinate and a velocity vector, where a fluid only has a velocity vector field (the continuity equation of a fluid in some way corresponds to the space coordinate of particles). In the code particles are present either as tracer particles or as dust particles
2.6.18.1. Tracer particles
Tracer particles always have the local velocity of the gas. The dynamical equations are thus
where the index \(i\) runs over all particles. Here \(\vec{u}\) is the gas velocity
at the position of the particle. One can choose between a first order (default)
and a second order spline interpolation scheme (set
lquadratic_interpolation=T in &particles_init_pars) to
calculate the gas velocity at the position of a tracer particle.
The sample run samples/dust-vortex contains the latest setup for
tracer particles.
2.6.18.2. Dust particles
Dust particles are allowed to have a velocity that is not similar to the gas,
The particle velocity follows an equation of motion similar to a fluid, only there is no advection term. Dust particles also experience a drag force from the gas (proportional to the velocity difference between a particle and the gas).
Here \(\tau_{\rm s}\) is the stopping time of the dust particle. The interpolation of the gas velocity to the position of a particle is done using one of three possible particle-mesh schemes,
- NGP (Nearest Grid Point, default)
The gas velocity at the nearest grid point is used.
- CIC (Cloud in Cell, set
lparticlemesh_cic=T) A first order interpolation is used to obtain the gas velocity field at the position of a particle. Affects 8 grid points.
- CIC (Cloud in Cell, set
- TSC (Triangular Shaped Cloud, set
lparticlemesh_tsc=T) A second order spline interpolation is used to obtain the gas velocity field at the position of a particle. Affects 27 grid points.
- TSC (Triangular Shaped Cloud, set
The particle description is the proper description of dust grains, since they do not feel any pressure forces (too low number density). Thus there is no guarantee that the grains present within a given volume will be equilibrated with each other, although drag force may work for small grains to achieve that. Larger grains (meter-sized in protoplanetary discs) must be treated as individual particles.
To conserve momentum the dust particles must affect the gas with a friction force as well. The strength of this force depends on the dust-to-gas ratio \(\epsilon_{\rm d}\), and it can be safely ignored when there is much more gas than there is dust, e.g., when \(\epsilon_{\rm d}=0.01\). The friction force on the gas appears in the equation of motion as
Here \(\rho_{\rm p}^{(i)}\) is the dust density that particle \(i\) represents.
This can be set through the parameter eps_todt in
&particle_init_pars. The drag force is assigned from the particles onto
the mesh using either NGP, CIC or TSC assignment. The same scheme is used both
for interpolation and for assignment to avoid any risk of a particle
accelerating itself (see Hockney & Eastwood 1981).
2.6.19. $N$-body solver
The \(N\)-body code takes advantage of the existing Particles module, which was coded with the initial intent of treating solid particles whose radius \(a_\bullet\) is comparable to the mean free path \(\lambda\) of the gas, for which a fluid description is not valid. A \(N\)-body implementation based on that module only needed to include mass as extra state for the particles, solve for the \(N^2\) gravitational pair interactions and distinguish between the \(N\)-body and the small bodies that are mapped into the grid as a \(\rho_p\) density field.
The particles of the \(N\)-body ensemble evolve due to their mutual gravity and by interacting with the gas and the swarm of small bodies. The equation of motion for particle \(i\) is
where \({\mathcal R}_{ij}=|\vec{r}_{p_i}-{\vec{r}}_{p_j}|\) is the distance between particles \(i\) and \(j\), and \(\hat{\vec{{\mathcal R}}}_{ij}\) is the unit vector pointing from particle \(j\) to particle \(i\). The first term of the R.H.S. is the combined gravity of the gas and of the dust particles onto the particle \(i\), solved via
where the integration is carried out over the whole disk. The smoothing distance \(b_i\) is taken to be as small as possible (a few grid cells). For few particles (\(<10\)), calculating the integral for every particle is practical. For larger ensembles one would prefer to solve the Poisson equation to calculate their combined gravitational potential.
The evolution of the particles is done with the same third-order Runge-Kutta time-stepping routine used for the gas. The particles define the timestep also by the Courant condition that they should not move more than one cell at a time. For pure particle runs, where the grid is absent, one can adopt a fixed time-step \(t_p \ll 2\pi\varOmega_{\rm fp}^{-1}\) where \(\varOmega_{\rm fp}\) is the angular frequency of the fastest particle.
By now (spring 2009), no inertial accelerations are included in the \(N\)-body module, so only the inertial frame - with origin at the barycenter of the \(N\)-body ensemble - is available. For a simulation of the circular restricted three-body problem with mass ratio \(q=\ttimes{-3}\), the Jacobi constant of a test particle initially placed at position \((x,y)=(2,0)\) was found to be conserved up to one part in \(\ttimes{5}\) within the time span of 100 orbits.
We stress that the level of conservation is poor when compared to integrators designed to specifically deal with long-term \(N\)-body problems. These integrators are usually symplectic, unlike the Runge-Kutta scheme of the PENCIL CODE. As such, PENCIL CODE should not be used to deal with evolution over millions of years. But for the time-span typical of astrophysical hydrodynamical simulations, this degree of conservation of the Jacobi constant can be deemed acceptable.
As an extension of the particle’s module, the \(N\)-body is fully compatible
with the parallel optimization of Pencil, which further speeds up the
calculations. Parallelization, however, is not yet possible for pure particle
runs, since it relies on splitting the grid between the processors. At the
time of writing (spring 2009), the \(N\)-body code does not allow the particles
to have a time-evolving mass.
The module pointmasses.f90 is also an \(N\)-body solver.
2.6.20. Test-field equations
The test-field method is used to calculate turbulent transport coefficients for magnetohydrodynamics. This is a rapidly evolving field and we refer the interested reader to recent papers in this field, e.g., by Sur et al. (2008) or Brandenburg et al. (2008). For technical details; see also Sect.~ref{RestartingFromLessPhysics}.
2.6.21. Gravitational waves equations
The expansion of the universe with time is described by the scale factor \(a(\tau)\), where \(\tau\) is the physical time. Using conformal time,
and dependent variables that are appropriately scaled with powers of \(a\), the hydromagnetic equations can be expressed completely without scale factor [BEO96,Dur08]_. This is not true, however, for the gravitational wave (GW) equations, where a dependence on \(a\) remains [Dur08]. The time dependence of \(a\) can be modeled as a power law, \(a\propto\tau^n\), where \(n=1/2\) applies to the radiation-dominated era; see Table 2.5 for the general relationship. To compare with cases where the expansion is ignored, we put \(n=0\).
\(n\) |
\(a\) |
\({\cal H}\) |
\(H\) |
|---|---|---|---|
\(0\) |
\(1\) |
\(0\) |
\(0\) |
\(1/2\) |
\(\eta/2\) |
\(1/\eta\) |
\(1/\eta\) |
\(2/3\) |
\(\eta^2/3\) |
\(2/\eta\) |
\(6/\eta^2\) |
In the transverse traceless (TT) gauge, the six components of the spatial part of the symmetric tensor characterizing the linearized evolution of the metric perturbations \(h_{ij}\) reduce to two components which, in the linear polarization basis, are the \(+\) and \(\times\) polarizations. However, the projection onto that basis is computationally intensive because it requires nonlocal operations involving Fourier transformations. It is therefore advantageous to evolve instead the perturbation of the metric tensor, \(h_{ij}\), in an arbitrary gauge, compute then \(h_{ij}^{\rm TT}\) in the TT gauge, and perform the decomposition into the linear polarization basis whenever diagnostic quantities such as averages or spectra are computed. Thus, we solve the linearized GW equation in the form [Dur08]:
for the six components \(1 \le i \le j \le 3\), where \(t\) is comoving time, \(a\) is the scale factor, \({\cal H} = \dot{a}/a\) is the comoving Hubble parameter, \(T_{ij}\) is the source term, \(c\) is the speed of light, and \(G\) is Newton’s constant. For \(n=0\), when the cosmic expansion is ignored, we have \(a=1\) and \({\cal H}=0\). In practice, we solve the GW equation for the scaled variable \(\tilde{h}_{ij} = a h_{ij}\):
For the numerical treatment of (2.29) or (2.30) and equations (2.32) and (2.34).
The source term is chosen to be the traceless part of the stress tensor:
The removal of the trace is not strictly necessary, but it prevents a continuous build-up of a large trace, which would be numerically disadvantageous. Viscous stress is ignored as it is usually small.
We compute \(T_{ij}\) by solving the energy, momentum, and induction equations for an ultrarelativistic gas [BEO96],[BKMRPTV17]_:
where \(\Bv = \nabla \times \Av\) is the magnetic field expressed in terms of the vector potential to ensure \(\nab \cdot \Bv = 0\), \(\Jv = \nab \times \Bv\) is the current density, \(\DD / \DD t = \partial / \partial t + \uv \cdot \nab\) is the advective derivative, \(\SSSS_{ij} = \frac{1}{2}(u_{i,j} + u_{j,i}) - \frac{1}{3} \delta_{ij} u_{k,k}\) is the trace-free rate-of-strain tensor, and \(p = \rho \cs^2\) is the pressure, where \(\cs = c / \sqrt{3}\) is the sound speed for an ultra-relativistic gas. Lorentz-Heaviside units are used for the magnetic field.
We are interested in the RMS value of the metric tensor perturbations and the GW energy density in the linear polarization basis. To compute \(h_{ij}^{\rm TT}\) from \(h_{ij}\), we Fourier transform the six components of \(h_{ij}\) and \(\dot{h}_{ij}\):
and compute the components in the TT gauge as
where \(P_{ij} = \delta_{ij} - \hat{k}_i \hat{k}_j\) is the projection operator, and \(\hat{\kv} = \kv/k\) is the unit vector of \(\kv\), with \(k = |\kv|\) being the modulus. Next, we compute the linear polarization bases
where \(e^1\) and \(e^2\) are unit vectors perpendicular to \(\kv\). Then
Returning to real space, we have
Analogous calculations are performed for \(\dot{h}_{+/\times}(\xv,t)\), which are used to compute the GW energy:
where angle brackets denote volume averages.
Fig. 2.5 Scalings of the relative error in the magnetic field, \((\delta B)_{\rm rms}/B_{\rm rms}\), and the gravitational strain \((\delta h)_{\rm rms}/h_{\rm rms}\) for GWs generated by the chiral magnetic effect, which leads to an exponentially increasing magnetic field. Low-resolution (\(32^3\)) versions of the Runs B4, B7, and B10 of [BHKRS21].
Analogous to kinetic and magnetic energy and helicity spectra, it is convenient to compute the GW energy and polarization spectra integrated over concentric shells of surface \(\int_{4\pi} k^2 d\Omega_\kv\) in \(\kv\) space:
The spectra are normalized such that \(\int_0^\infty \Shdot(k) \, dk = \langle \dhT^2 \rangle + \langle \dhX^2 \rangle\) is proportional to the energy density, and \(\int_0^\infty \Ahdot(k) \, dk\) is proportional to the polarized energy density. The \(\Ahdot(k)\) spectra are not to be confused with the magnetic vector potential \(\Av(\xv,t)\).
The corresponding GW energy spectra are:
We also define spectra for the metric tensor perturbation:
normalized such that \(\int_0^\infty \Sh(k) \, dk = \hrms^2\) is the mean squared metric tensor perturbation.
Assuming the GW source is constant between two time steps gives an accuracy that scales linearly with the time step \(\delta t\), while the magnetic field, related to the magnetic stress, scales cubically with \(\delta t\). Since \(\tilde{T}_{+/\times}\) are stored in the f-array, it is easy to extract for each wavevector the increment of the stress \(\delta \tilde{T}_{+/\times}\) and use it in an improved update of the GW field:
This more accurate solver is invoked by setting itorder_GW=2, which results in a quadratic scaling of the error of the GW field; see Fig. 2.5.
2.7. Troubleshooting / Frequently Asked Questions
2.7.1. Download and setup
2.7.1.1. Download forbidden
http://de.wikipedia.org/wiki/GitHub
http://en.wikipedia.org/wiki/SourceForge
A: Both Github and SourceForge are banned from countries on the United States Office of Foreign Assets Control sanction list, including Cuba, Iran, Libya, North Korea, Sudan and Syria; see http://de.wikipedia.org/wiki/GitHub and http://en.wikipedia.org/wiki/SourceForge . As a remedy, you might download a tarball from https://pencil-code.org/. See also Section Download the Pencil Code.
2.7.1.2. When sourcing the sourceme.sh/sourceme.csh file or running pc_setupsrc, I get error messages from the shell, like if: Expression Syntax. or set: Variable name must begin with a letter.
A: This sounds like a buggy shell setup, either by yourself or your system administrator — or a shell that is even more idiosyncratic than the ones we have been working with.
To better diagnose the problem, collect the following information before filing a bug report to us:
uname -a/bin/csh -vecho $versionecho $SHELLps -p $$If you have problems while sourcing the
sourcemescript:Unset the
PENCIL_HOMEvariable:for
cshand similar:unsetenv PENCIL_HOMEfor
bashand similar:unexport PENCIL_HOME; unset PENCIL_HOME
Switch your shell in verbose mode:
for
cshand similar:set verbose; set echofor
bashand similar:set -v; set -xthen source again.
If you have problems with pc_setupsrc, run it with
cshin verbose mode:/bin/csh -v -x $PENCIL_HOME/bin/pc_setupsrc
2.7.2. Compilation
2.7.2.1. Error: relocation truncated to fit
If you get errors while compiling and linking that are similar to:
density.f90:(.text+0x5e0): relocation truncated to fit: R_X86_64_PC32
against symbol `cdata_mp_m_' defined in COMMON section in cdata.o
density.f90:(.text+0x644): additional relocation overflows omitted from the
output
make[2]: *** [start.x] Error 1
A: Your setup is probably too large to fit a normal’ memory model.
Please choose a `medium’ or `large’ memory model by adding one of these
compiler options to your configuration: :command:-mcmodel=medium` or -mcmodel=large.
See Sect. Configuring the code to compile and run on your computer for configuration details. Alternatively,
if you use pc_build, you may simply add the respective extension:
pc_build -f GNU-GCC_MPI,GNU-GCC_medium
or for the Intel compiler and a large memory model you would use:
pc_build -f Intel_MPI,Intel_large
2.7.2.2. Linker can’t find the syscalls functions:
ld: 0711-317 ERROR: Undefined symbol: .is_nan_c
ld: 0711-317 ERROR: Undefined symbol: .sizeof_real_c
ld: 0711-317 ERROR: Undefined symbol: .system_c
ld: 0711-317 ERROR: Undefined symbol: .get_env_var_c
ld: 0711-317 ERROR: Undefined symbol: .get_pid_c
ld: 0711-317 ERROR: Undefined symbol: .file_size_c
A: The Pencil Code needs a working combination of a Fortran- and a
C-compiler. If this is not correctly set up, usually the linker won’t find
the functions inside the syscalls module. If that happens, either the
combination of C- and Fortran-compiler is inappropriate (e.g., ifort
needs icc), or the compiler needs additional flags, like g95
might need the option -fno-second-underscore and xlf might
need the option -qextname.
Please refer to Sect. Experimenting with compiler flags, Table Compiler flags for common compilers.
2.7.2.3. Make gives the following error now:
PGF90-S-0017-Unable to open include file: chemistry.h (nochemistry.f90: 43)
0 inform, 0 warnings, 1 severes, 0 fatal for chemistry
Line 43 of the nochemistry routine, only has 'contains'.
A: This is because somebody added a new module (together with a
corresponding nomodule.f90 and a module.h file (chemistry in this
case). These files didn’t exist before, so you need to say:
pc_setupsrc
If this does not help, say first make clean and then pc_setupsrc.
2.7.2.4. How do I compile the Pencil Code with the Intel (ifc) compiler under Linux?
A: The Pencil Code should compile successfully with ifc``~6.x,
``ifc``~7.0, sufficiently recent versions of ``ifc 7.1 (you
should get the latest version; if yours is too old, you will typically
get an internal compiler error’ during compilation of
``src/hydro.f90`), as well as with recent versions of
ifort 8.1 (8.0 may also work).
You can find the ifort compiler at
<ftp://download.intel.com/software/products/compilers/downloads>_.
On many current (as of November 2003) Linux systems, there is a mismatch
between the glibc versions used by the compiler and the linker.
To work around this, use the following flag for compiling
FC=ifc -i_dynamic
and set the environment variable
LD_ASSUME_KERNEL=2.4.1; export LD_ASSUME_KERNEL
or
setenv LD_ASSUME_KERNEL 2.4.1
This has solved the problems e.g., on a system with glibc-2.3.2 and kernel 2.4.22.
Thanks to Leonardo J. Milano (http://udel.edu/~lmilano/) for part of this info.
2.7.2.5. I keep getting segmentation faults with start.x when compiling with ifort 8.
A: There was/is a number of issues with ifort 8.0.
Make sure you have the latest patches applied to the compiler.
A number of things to consider or try are:
Compile with the the -static -nothreads flags.
Set your stacksize to a large value (but a far too large value may be problematic, too), e.g.
limit stacksize 256m
ulimit -s 256000
Set the environment variable KMP_STACKSIZE to a large value (like
100M)
See also http://softwareforums.intel.com/ids/board/message?board.id=11&message.id=1375
2.7.2.6. When compiling with MPI on a Linux system, the linker complains:
mpicomm.o: In function `mpicomm_mpicomm_init_':
mpicomm.o(.text+0x36): undefined reference to `mpi_init_'
mpicomm.o(.text+0x55): undefined reference to `mpi_comm_size_'
mpicomm.o(.text+0x6f): undefined reference to `mpi_comm_rank_'
[...]
A: This is the infamous underscore problem.
Your MPI libraries have been compiled with G77 without the
option -fno-second-underscore, which makes the MPI
symbol names incompatible with other Fortran compilers.
As a workaround, use
MPICOMM = mpicomm_
in Makefile.local.
Or, even better, you can set this globally (for the given computer) by
inserting that line into the file
~/.adapt-mkfile.inc
(see perldoc adapt-mkfile for more details).
2.7.2.7. Compilation stops with the cryptic error message:
f95 -O3 -u -c .f90.f90
Error : Could not open sourcefile .f90.f90
compilation aborted for .f90.f90 (code 1)
make[1]: *** [.f90.o] Error 1
What is the problem?
A: There are two possibilities:
One of the variables for
makehas not been set, somakeexpands it to the empty string. Most probably you forgot to specify a module insrc/Makefile.local. One possibility is that you have upgraded from an older version of the code that did not have some of the modules the new version has.Compare your
src/Makefile.localto one of the examples that work.One of the variables for
makehas a space appended to it, e.g., if you use the line
MPICOMM = mpicomm_
see When compiling with MPI on a Linux system, the linker complains:. With a trailing blank, you will encounter this error message.
Remove the blank.
This problem can also occur if you added a new module (and have an empty space after the module name in
src/Makefile.src, i.e.CHIRAL=nochiral `), in which case the compiler will talk about ``circular dependence`for the filenochiral.
2.7.2.8. The code doesn’t compile,
there is a problem with mvar:
make start.x run.x
f95 -O3 -u -c cdata.f90
Error: cdata.f90, line 71: Implicit type for MVAR
detected at MVAR@)
[f95 terminated - errors found by pass 1]
make[1]: *** [cdata.o] Error 2
A: Check and make sure that mkcparam (directory
$PENCIL_HOME/bin) is in your path.
If this doesn’t help, there may be an empty cparam.inc
file in your src/ directory.
Remove cparam.inc and try again
(Note that cparam.inc is automatically generated from the
Makefile).
2.7.2.9. Some samples don’t even compile,
As you can see on the web, http://www.nordita.org/software/pencil-code/tests.html.
samples/helical-MHDturb:
Compiling.. not ok:
make start.x run.x read_videofiles.x
make[1]: Entering directory `/home/dobler/f90/pencil-code/samples/helical-MHDturb/src'
/usr/lib/lam/bin/mpif95 -O3 -c initcond.f90
/usr/lib/lam/bin/mpif95 -O3 -c density.f90
use Gravity, only: gravz, nu_epicycle
^
Error 208 at (467:density.f90) : No such entity in the module
Error 355 : In procedure INIT_LNRHO variable NU_EPICYCLE has not been given a type
Error 355 : In procedure POLYTROPIC_LNRHO_DISC variable NU_EPICYCLE has not been given a type
3 Errors
compilation aborted for density.f90 (code 1)
make[1]: *** [density.o] Error 1
make[1]: Leaving directory `/home/dobler/f90/pencil-code/samples/helical-MHDturb/src'
make: *** [code] Error 2
A: Somebody may have checked in something without having
run auto-test beforehand. The problem here is that something has been added in
one module, but not in the corresponding no-module. You can of course check
with svn who it was…
2.7.2.10. Internal compiler error with Compaq/Dec F90
The Dec Fortran optimizer has occasional problems with nompicomm.f90:
make start.x run.x read_videofiles.x
f90 -fast -O3 -tune ev6 -arch ev6 -c cparam.f90
[...]
f90 -fast -O3 -tune ev6 -arch ev6 -c nompicomm.f90
otal vm 2755568 otal vm 2765296 otal vm 2775024
otal vm 2784752 otal...
Assertion failure: Compiler internal error - please submit problem r...
GEM ASSERTION, Compiler internal error - please submit problem report
Fatal error in: /usr/lib/cmplrs/fort90_540/decfort90 Terminated
*** Exit 3
Stop.
*** Exit 1
Stop.
A: The occurrence of this problem depends upon the grid size;
and the problem never seems to occur with mpicomm.f90,
except when ncpus=1.
The problem can be avoided by switching off the loop transformation
optimization (part of the -O3 optimization), via:
#OPTFLAGS=-fast -O3 -notransform_loops
This is currently the default compiler setting in Makefile,
although it has a measurable performance impact (some 8% slowdown).
2.7.2.11. Assertion failure under SunOS
Under SunOS, I get an error message like
user@sun> f90 -c param_io.f90
Assertion failed: at_handle_table[at_idx].tag == VAR_TAG,
file ../srcfw/FWcvrt.c, line 4018
f90: Fatal error in f90comp: Abort
A: This is a compiler bug that we find at least with Sun’s WorkShop Compiler version 5.0 00/05/17 FORTRAN 90 2.0 Patch 107356-05. Upgrade the compiler version (and possibly also the operating system): we find that the code compiles and works with version Sun WorkShop 6 update 2 Fortran 95 6.2 Patch 111690-05 2002/01/17 under SunOS version 5.8 Generic_108528-11.
2.7.2.12. After some dirty tricks I got pencil code to compile with MPI, …
> Before that i installed lam-7.1.4 from source.
Goodness gracious me, you shouldn't have to compile your own MPI library.
A: Then don’t use the old LAM-MPI. It is long superseded by open-mpi now. Open-mpi doesn’t need a daemon to be running. I am using the version that ships with Ubuntu (e.g., 9.04):
frenesi:~> aptitude -w 210 search openmpi | grep '^i'
i libopenmpi-dev - high performance message passing library -- header files
i A libopenmpi1 - high performance message passing library -- shared library
i openmpi-bin - high performance message passing library -- binaries
i A openmpi-common - high performance message passing library -- common files
i openmpi-doc - high performance message passing library -- man pages
Install that and keep your configuration (Makefile.src and getconf.csh) close to that for frenesi or norlx50. That should work.
2.7.2.13. Error: Symbol ‘mpi_comm_world’ at (1) has no IMPLICIT type
I installed the pencil code on Ubuntu system and tested "run.csh"
in ...\samples\conv-slab. Here the code worked pretty well.
Nevertheless, running (auto-test), I found there are some errors.
The messages are,
Error: Symbol 'mpi_comm_world' at (1) has no IMPLICIT type
Fatal Error: Error count reached limit of 25.
make[2]: *** [mpicomm_double.o] Error 1
make[2]: Leaving directory
`/home/pkiwan/Desktop/pencil-code/samples/2d-tests/selfgravitating-shearwave/src'
make[1]: *** [code] Error 2
make[1]: Leaving directory
`/home/pkiwan/Desktop/pencil-code/samples/2d-tests/selfgravitating-shearwave/src'
make: *** [default] Error 2
Finally, ### auto-test failed ###
Will it be OK? Or, how can I fix this?
A: Thanks for letting me know about the status, and congratulations on your progress! Those tests that fail are those that use MPI. If your machine is a dual or multi core machine, you could run faster by running under MPI. But this is probably not crucial for you at this point. (I just noticed that there is a ToDo listed in the auto-test command to implement the option not to run the MPI tests, but this hasn’t been done yet. So I guess you can start with the science next.
2.7.2.14. Error: Can’t open included file ‘mpif.h’
It always worked, but now, after some systems upgrade, I get
gfortran -O3 -o mpicomm.o -c mpicomm.f90
Error: Can't open included file 'mpif.h'
When I say locate mpif.h I only get things like
/scratch/ntest/1.2.7p1-intel/include/mpif.h
But since I use FC=mpif90 I thought I don’t need to worry.
A: Since you use FC=mpif90 there must definitely be
something wrong with their setup.
Try mpif90 -showme or mpif90 -show; the -I` option
should say where it looks for ‘mpif.h’.
If those directories don’t exist, it’s no wonder that it doesn’t work,
and it is time to complain.
2.7.2.15. Compilation fails on MacOS Sonoma or Monterey
I am getting an error which hasn’t been resolved yet. It has something to do with my Mac setup and no one has been able to figure out how to fix it. Maybe you’ve seen this before (at the pc_build stage):
'make -j FFLAGS_DOUBLE=-fdefault-real-8 -fdefault-double-8
CFLAGS_DOUBLE=-DDOUBLE_PRECISION LD_MPI= CFLAGS_FFTW3= FFLAGS_FFTW3=
LD_FFTW3= CFLAGS_FFTW2= FFLAGS_FFTW2= LD_FFTW2= FC=gfortran F77=$(FC)
FFLAGS=-O LDFLAGS_HELPER=-dynamic OMPFFLAGS=-fopenmp OMPLFLAGS=-lgomp
PPFLAGS=-cpp FSTD_95=-std=f95 FSTD_2003=-std=f2003 CC=gcc
CFLAGS=-DFUNDERSC=1 default_to_be' failed: <Inappropriate ioctl for device>
A: You have to use gcc-14. Default (plain gcc) does not work; it is wrongly set up.
2.7.2.16. Compilation fails on Tanmay’s MacOS
In my case the gcc compiler was renamed gcc-14. Once this was fixed, all was good. Could it be that when Mac updates its OS it changes the name of the compiler? I don’t know the backend of the Mac so well.
2.7.2.17. Missing ld_classic on MacOS
While running pc_build, I get an error message saying that -ld_classic is not found.
A:
Note that ld_classic is not specifying a library called libd_classic, but is rather an option passed to Apple’s ld binary.
type ld should say /usr/bin/ld.
This error has been noticed on systems where people tried to install gfortran using anaconda, which pulls in a wrongly configured ld binary.
Run conda remove ld64 and use another method, like homebrew, to install gfortran.
2.7.2.18. Further MacOS tips
For further MacOS tips, see also the “Instructions for MacOS installation” on http://pencil-code.nordita.org/doc.php.
2.7.3. Pencil check
2.7.3.1. The pencil check complains for no reason.
A: The pencil check only complains for a reason.
2.7.3.2. The pencil check reports MISSING PENCILS and quits
A: This could point to a serious problem in the code. Check where the
missing pencil is used in the code. Request the right pencils, likely based on
input parameters, by adapting one or more of the pencil_criteria_MODULE
subroutines.
2.7.3.3. The pencil check reports unnecessary pencils
The pencil check reports
possible overcalculation... pencil rho ( 43) is requested, but does not
appear to be required!
A: Such warnings show that your simulation is possibly running too slowly
because it is calculating pencils that are not actually needed. Check in the
code where the unnecessary pencils are used and adapt one or more of the
pencil_criteria_MODULE subroutines to request pencils only when they are
actually needed.
2.7.3.4. The pencil check reports that most or all pencils are missing
A: This is typically a thing that can happen when testing new code
development for the first time. It is usually an indication that the reference
df changes every time you call pde. Check whether any newly
implemented subroutines or functionality has a “memory”, i.e. if calling the
subroutine twice with the same f gives different output df.
2.7.3.5. Running the pencil check triggers mathematical errors in the code
A: The pencil check puts random numbers in f before checking the
dependence of df on the chosen set of pencils. Sometimes these random
numbers are inconsistent with the physics and cause errors. In that case you
can set lrandom_f_pencil_check=F in &run_pars in run.in.
The initial condition may contain many idealized states (zeros or ones) which
then do not trigger pencil check errors when lrandom_f_pencil_check=F,
even if pencils are missing. But it does prevent mathematical inconsistencies.
2.7.3.6. The pencil check still complains
A: Then you need to look into the how the code and the pencil check
operate. Reduce the problem in size and dimensions to find the smallest
problem that makes the pencil check fail (e.g., 1x1x8 grid points). At the line
of pencil_check.f90 when a difference is found between df_ref and
df, add some debug lines telling you which variable is inconsistent and
in what place. Often you will be surprised that the pencil check has correctly
found a problem in the simulation.
2.7.3.7. The pencil check is annoying so I turned it off
A: Then you are taking a major risk. If one or more pencils are not calculated properly, then the results will be wrong.
2.7.4. Running
2.7.4.1. Why does start.x / start.csh write data with periodic boundary conditions?
A: Because you are setting the boundary conditions in run.in, not
in start.in; see Sect. Where to specify boundary conditions. There is nothing wrong
with the initial data — the ghost-zone values will be re-calculated during
the very first time step.
2.7.4.2. csh problem?
Q: On some rare occasions we have problems with csh not being supported on other machines. (We hope to fix this by contacting the responsible person, but may not be that trivial today!) Oliver says this is a well known bug of some years ago, etc. But maybe in the long run it would be good to avoid csh.
A: These occasions will become increasingly frequent, and eventually for some architectures, there may not even be a csh variant that can be installed.
We never pushed people to use pc_run and friends (and to report corresponding bugs and get them fixed), but if we don’t spend a bit of effort (or annoy users) now, we create a future emergency, where someone needs to run on some machine, but there is no csh and he or she just gets stuck.
We don’t have that many csh files, and for years now it should be possible
to compile run without csh (using bin/pc_run) — except that people
still fall back on the old way of doing things.
This is both cause and consequence of the new way not being tested that
much, at least for the corner cases like RERUN, NEWDIR,
SCRATCH_DIR.
2.7.4.3. run.csh doesn’t work:
Invalid character ''' in NAMELIST input
Program terminated by fatal I/O error
Abort
A: The string array for the boundary condition, e.g., bcx or
bcz is too long. Make sure it has exactly as many elements as nvar
is big.
2.7.4.4. Code crashes after restarting
> > removing mu_r from the namelist just `like that' makes the code
> > backwards incompatible.
>
> That means that we can never get rid of a parameter in start.in once we
> have introduced it, right?
A: In the current implementation, without a corresponding cleaning procedure, unfortunately yes.
Of course, this does not affect users’ private changes outside the central svn
tree.
2.7.4.5. auto-test gone mad…?
Q: Have you ever seen this before:
giga01:/home/pg/n7026413/cvs-src/pencil-code/samples/conv-slab> auto-test
.
/home/pg/n7026413/cvs-src/pencil-code/samples/conv-slab:
Compiling.. ok
No data directory; generating data -> /var/tmp/pencil-tmp-25318
Starting.. ok
Running.. ok
Validating results..Malformed UTF-8 character (unexpected continuation
byte 0x80, with no preceding start byte) in split at
/home/pg/n7026413/cvs-src/pencil-code/bin/auto-test line 263.
Malformed UTF-8 character (unexpected continuation byte 0x80, with no
preceding start byte) in split at
/home/pg/n7026413/cvs-src/pencil-code/bin/auto-test line 263.
A: You are running on a RedHat 8 or 9 system, right?
Set LANG=POSIX in your shell’s startup script and life will be
much better.
2.7.4.6. Can I restart with a different number of cpus?
Q: I am running a simulation of nonhelical turbulence on the cluster using MPI. Suppose if I am running a \(128^3\) simulation on 32 cpus/cores i.e.
integer, parameter :: ncpus=32,nprocy=2,nprocz=ncpus/nprocy,nprocx=1
integer, parameter :: nxgrid=128,nygrid=nxgrid,nzgrid=nxgrid
And I stop the run after a bit. Is there a way to resume this run with different number of cpus like this :
integer, parameter :: ncpus=16,nprocy=2,nprocz=ncpus/nprocy,nprocx=1
integer, parameter :: nxgrid=128,nygrid=nxgrid,nzgrid=nxgrid
I understand it has to be so in a new directory but making sure that the run starts from where I left it off in the previous directory.
A: The answer is no, if you use the standard distributed io. There is also parallel io, but I never used it. That would write the data in a single file, and then you could use the data for restart in another processor layout.
2.7.4.7. Can I restart with a different number of cpus?
Q:
Is it right that once the simulation is resumed, pencil-code takes the
last data from var.dat (which is the current snapshot of the fields)?
If that is true, then, is it not possible to give that as the initial
condition for the run in the second directory (with changed "ncpus")?
Is there a mechanism already in place for that?
A:
Yes, the code restarts from the last var.dat. It is written after a
successful completion of the run, but it crashes or you hit a time-out,
there will be a var.dat that is overwritten every isave timesteps.
If the system stops during writing, some var.dat files may be corrupt
or have the wrong time. In that case you could restart from a good VAR
file, if you have one, using, e.g.,
restart-new-dir-VAR . 46
where 46 is the number of your VAR file, i.e., VAR46 im this case. To restart in another directory, you say, from the old run directory,
restart-new-dir ../another_directory
Hope this helps.
Look into pencil-code/bin/restart-new-dir to see what it is doing.
2.7.4.8. fft_xyz_parallel_3D: nygrid needs to be an integer multiple…
Q: I just got an:
fft_xyz_parallel_3D: nygrid needs to be an integer multiple of nprocy*nprocz
In my case, nygrid=2048, nprocy=32, and nprocz=128, so nprocy*nprocz=4096. In other words, 2048 needs to be a multiple of 4096. But isn’t this the case then?
A: No, because 2048 = 0.5 * 4096 and 0.5 is not an integer. Maybe try either setting nprocz=64 or nprocy=64. You could compensate the change of ncpus with the \(x\)-direction. For \(2048^3\) simulations, nprocy=32 and nprocz=64 would be good. A list of good meshes is given in Table 2.4.
2.7.4.9. Unit-agnostic calculations?
Q:
The manual speaks about unit-agnostic calculations, stating that one may
choose to interpret the results in any (consistent) units, depending
on the problem that is solved at hand. So, for example, if I chose to
run the 2d-tests/battery_term simulation for an arbitrary number of
time-steps and then choose to examine the diagnostics, am I correct in
assuming the following:
1) [Brms] = Gauss (as output by unit_magnetic, before the run begins)
2) [t] = s (since the default unit system is left as CGS)
3) [urms] = cm/s (again, as output by unit_velocity, before the run begins)
4) and etc. for the units of the other diagnostics
A: Detailed correspondence on this item can be found on: <https://groups.google.com/forum/?fromgroups#!topic/pencil-code-discuss/zek-uYNbgXI>_ There is also working material on unit systems under <http://www.nordita.org/~brandenb/teach/PencilCode/MixedTopics.html>_ with a link to <http://www.nordita.org/~brandenb/teach/PencilCode/material/AlfvenWave_SIunits/>_ Below is a pedagogical response from Wlad Lyra:
In the sample battery-term, the sound speed cs0=1 sets the unit of
velocity. Together with the unit of length, that sets your unit of
time. The unit of magnetic field follows from the unit of velocity,
density, and your choice of magnetic permittivity, according to the
definition of the Alfven velocity.
If you are assuming cgs, you are saying that your sound speed cs0=1
actually means [U]=1 cm/s. Your unit of length is equivalently 1 cm,
and therefore the unit of time is [t] = [L]/[U]=1 s. The unit of
density is [rho] = 1 g/cm^3. Since in cgs vA=B/sqrt(4*pi * rho), your
unit of magnetic field is [B] = [U] * sqrt([rho] * 4*pi) ~= 3.5
sqrt(g/cm) / s = 3.5 Gauss.
If instead you are assuming SI, you have cs0=1 assuming that means
[U]=1 m/s and rho0=1 assuming that to mean [rho]=1 kg/m^3. Using [L]=1
m, you have still [t]=1 s, but now what appears as B=1 in your output
is actually [B] = [U] * sqrt (mu * [rho]) = 1 m/s * sqrt(4*pi * 1e-7
N*A-2 1 kg/m^3) ~= 0.0011210 kg/(s^2*A) ~ 11 Gauss.
You can make it more interesting and use units relevant to the
problem. Say you are at the photosphere of the Sun. You may want to
use dimensionless cs0=1 meaning a sound speed of 10 km/s. Your
appropriate length can be a megameter. Now your time unit is
[t]=[L]/[U] = 1e3 km/ 10 km/s = 10^2 s, i.e., roughly 1.5 minute. For
density, assume rho=2x10-4 kg/m^3, typical of the solar photosphere.
Your unit of magnetic field is therefore [B] = [U] * sqrt([rho] *
4*pi) = 1e6 cm/s * sqrt(4*pi * 2e-7 g/cm^3) ~ 1585.33 Gauss.
Notice that for mu0=1 and rho0=1 you simply have vA=B. Then you can
conveniently set the field strength by your choice of plasma beta (=
2*cs^2/vA^2). There's a reason why we like dimensionless quantities!
2.7.5. Visualization FAQ
2.7.5.1. start.pro doesn’t work:
Reading grid.dat..
Reading param.nml..
% Expression must be a structure in this context: PAR.
% Execution halted at: $MAIN$ 104
/home/brandenb/pencil-code/runs/forced/hel1/../../../idl/start.pro
A: You don’t have the subdirectory data/ in your IDL variable !path. Make sure you source sourceme.csh/sourceme.sh or set a sufficient IDL path otherwise.
2.7.5.2. start.pro doesn’t work (2):
Isn’t there some clever (or even trivial) way that one can avoid the annoying error messages that one gets, when running e.g., .r rall after a new variable has been introduced in “idl/varcontent.pro”? Ever so often there’s a new variable that can’t be found in my param2.nml – this time it was IECR, IGG, and ILNTT that I had to circumvent…
A: The simplest solution is to invoke NOERASE, i.e. say
touch NOERASE
start.csh
or, alternatively, start_run.csh. What it does is that it reruns
src/start.x with a new version of the code; this then produces all
the necessary auxiliary files, but it doesn’t overwrite or erase the
var.dat and other VAR and slice files.
2.7.5.3. Something about tag name undefined:
Q: In one of my older run directories I can’t read the data with idl anymore. What should I do? It says something like
Reading param.nml..
% Tag name LEQUIDIST is undefined for structure <Anonymous>.
% Execution halted at: $MAIN$ 182
/people/disk2/brandenb/pencil-code/idl/start.pro
A: Go into data/param.nml and add , LEQUIDIST=T anywhere in
the file (but before the last slash).
2.7.5.4. Something INC in start.pro
Q: start doesn’t even work:
% Compiled module: $MAIN$.
nname= 11
Reading grid.dat..
Reading param.nml..
Can't locate Namelist.pm in INC (INC contains: /etc/perl /usr/local/lib/perl/5.8.4 /usr/local/share/perl/5.8.4 /usr/lib/perl5 /usr/share/perl5 /usr/lib/perl/5.8 /usr/share/perl/5.8 /usr/local/lib/site_perl .) at /home/brandenb/pencil-code/bin/nl2idl line 49.
BEGIN failed--compilation aborted at /home/brandenb/pencil-code/bin/nl2idl line 49.
A: Go into $PENCIL_HOME and say svn up sourceme.csh and/or
svn up sourceme.sh. (They were just out of date.)
2.7.5.5. nl2idl problem when reading param2.nml
Q: Does anybody encounter a backward problem with nl2idl? The file
param*.nml files are checked in under
pencil-code/axel/couette/SStrat128a_mu0.20_g2
and the problem is below.
at /people/disk2/brandenb/pencil-code/bin/nl2idl line 120
HCOND0= 0.0,HCOND1= 1.000000,HCOND2= 1.000000,WIDTHSS= 1.192093E-06,MPOLY0=
^------ HERE
at /people/disk2/brandenb/pencil-code/bin/nl2idl line 120
A: The problem is the ifc compiler writing the following into the namelist file:
COOLING_PROFILE='gaussian ',COOLTYPE='Temp
'COOL= 0.0,CS2COOL= 0.0,RCOOL= 1.000000,WCOOL= 0.1000000,FBOT= 0.0,CHI_T= 0.0
If you add a comma after the closing quote:
COOLING_PROFILE='gaussian ',COOLTYPE='Temp
',COOL= 0.0,CS2COOL= 0.0,RCOOL= 1.000000,WCOOL= 0.1000000,FBOT= 0.0,CHI_T= 0.0
things will work.
Note that ifc cannot even itself read what it is writing here, so if this happened to occur in param.nml, the code would require manual intervention after each start.csh.
2.7.5.6. Spurious dots in the time series file
Q: Wolfgang, you explained it to me once, but I forget. How can one remove spurious dots after the timestep number if the time format overflows?
A: I don’t know whether it exists anywhere, but it’s easy. In Perl you’d say
perl -pe 's/^(\s*[-0-9]+)\.([-0-9eEdD])/$1 $2/g'
and in sed (but that’s harder to read)
sed 's/^\( *[-0-9]\+\)\.\([-0-9eEdD]\)/\1 \2/g'
2.7.5.7. Problems with pc_varcontent.pro
Q:
% Subscript range values of the form low:high must be >= 0, < size, with low
<= high: VARCONTENT.
% Error occurred at: PC_VARCONTENT 391
/home/brandenb/pencil-code/idl/read/pc_varcontent.pro
% PC_READ_VAR 318
/home/brandenb/pencil-code/idl/read/pc_read_var.pro
% $MAIN$
A: Make sure you don’t have any unused items in your
src/cparam.local such as
! MAUX CONTRIBUTION 3
! COMMUNICATED AUXILIARIES 3
They would leave gaps in the counting of entries in your
data/index.pro file.
2.7.6. Programming new slices
Q: I’m creating a new special module with a few new variables f(:,:,:,inew1), f(:,:,:,inew2) etc.
I’m wondering how to add new output of slices (i.e. video files)
of these new variables (say p%inew1) and their combinations (say
p%inew1**2+p%inew2**2)? I tried to look into other modules to find an
example but got confused. If someone could clarify the calling tree it
would be great!
A: Best you look into hydro.f90. For slices, which contain simply f-array variables, you do something like
case ('uu'); call assign_slices_vec(slices,f,iuu)
in get_slices_hydro. For slices, which contain derived quantities like your
p%inew1**2+p%inew2**2, you have to declare slice buffers as for divu in hydro (divu_xy etc.), you have to allocate them like
if (ivid_divu/=0) call alloc_slice_buffers(divu_xy,divu_xz, &
divu_yz,divu_xy2,divu_xy3,divu_xy4,divu_xz2,divu_r)
to fill them in calc_diagnostics_special like
if (ivid_divu/=0) call store_slices(p%divu,divu_xy,divu_xz, &
divu_yz,divu_xy2,divu_xy3,divu_xy4,divu_xz2,divu_r)
and finally to store them in get_slices_special like
call assign_slices_scal(slices,divu_xy,divu_xz,divu_yz, &
divu_xy2,divu_xy3,divu_xy4,divu_xz2,divu_r)
2.7.7. General questions
2.7.7.1. Installation procedure
2.7.7.1.1. Why don’t you use GNU autoconf/automake for installation of the PENCIL CODE?
A: What do you mean by “installation”? Unlike the applications that normally use autoconf, the :Pencil Code: is neither a binary executable, nor a library that you compile once and then dump somewhere in the system tree. Autoconf is the right tool for these applications, but not for numerical codes, where the typical compilation and usage pattern is very different:
You have different directories with different Makefile.local settings,
recompile after introducing that shiny new term in your equations, etc.
Moreover, you want to sometimes switch to a different compiler (but just
for that run directory) or another MPI implementation. Our adapt-mkfile
approach gives you this flexibility in a reasonably convenient way, while
doing the same thing with autoconf would be using that system against most
of its design principles.
Besides, it would really get on my (WD’s) nerves if I had to wait two minutes for autoconf to finish before I can start compiling (or maybe 5–10 minutes if I worked on a NEC machine…).
Finally, if you have ever tried to figure out what a configure script
does, you will appreciate a comprehensible configuration system.
2.7.7.2. Small numbers in the code
What is actually the difference between epsi, tini and tiny?
A:
F90 has two functions epsilon() and tiny(), with
epsilon(x) = 1.1920929e-07
tiny(x) = 1.1754944e-38
(and then there is huge(x) = 3.4028235e+38)
for a single-precision number x.
epsilon(x) is the smallest number that satisfies
1+epsilon(1.) /= 1 ,
while tiny(x) is the smallest number that can be represented without
precision loss.
In the code we have variants hereof,
epsi=5*epsilon(1.0)
tini=5*tiny(1.0)
huge1=0.2*huge(1.0)
that have added safety margins, so we don't have to think about doing
things like 1/tini.
So in sub.f90,
- evr = evr / spread(r_mn+epsi,2,3)
did (minimally) affect the result for r_mn=O(1), while the correct version
+ evr = evr / spread(r_mn+tini,2,3)
only avoids overflow.
2.7.7.3. Why do we need a /lphysics/ namelist in the first place?
Wolfgang answered on 29 July 2010: “cdata.f90 has the explanation”
! Constant 'parameters' cannot occur in namelists, so in order to get the
! now constant module logicals into the lphysics name list...
! We have some proxies that are used to initialize private local variables
! called lhydro etc, in the lphysics namelist!
So the situation is this: we want to write parameters like ldensity to
param.nml so IDL (and potentially octave, python, etc.) can know whether
density was on or not. To avoid confusion, we want them to have exactly
their original names.
But we cannot assemble the original ldensity etc. constants in a
namelist, so we have to define a local ldensity variable. And to provide
it with the value of the original cdata.ldensity, we need to transfer the
value via ldensity_var.
That’s pretty scary, although it seems to work fine. I can track the
code back to the big eos_merger commit, so it may originate from that
branch.
One obvious problem is that you have to add code in a number of places
(the ldensity -> ldensity_var assignment and the local
definition of ldensity) to really get what you need.
And when adding a new boolean of that sort to cdata.f90,
you may not even have a clue that you need all the other voodoo.
2.7.7.4. Can I run the code on a Mac?
A: Macs work well for Linux stuff, except that the file structure is slightly different. Problems when following Linux installs can usually be traced to the PATH. For general reference, if you need to set an environment variable for an entire OS-X login session, google environment.plist. That won’t be needed here.
For a Mac install, the following should work:
Install Dev Tools (an optional install on the MacOS install disks). Unfortunately, last time I checked the svn version that comes with DevTools is obsolete. So:
Install MacPorts (download from web). Note that MacPorts installs to a non-standard location, and will need to be sourced. The installation normally drops an appropriate line in .profile. If it does so, make sure that line gets sourced. Otherwise
export PATH=/opt/local/bin:/opt/local/sbin:$PATH
export MANPATH=/opt/local/share/man:$MANPATH
Install g95 (download from web). Make sure it is linked in /bin.
execute macports svn install
download the pencil-code and enjoy.
Note
the above way to get svn works. It takes a while however, so there are certainly faster ways out there. If you already have a non-obsolete svn version, use that instead.
2.7.7.5. Wrong user-id in commit emails
Why does it say
From: 'Philippe Bourdin' via pencil-code-commits
<pencil-code-commits@googlegroups.com>
when the committing author is not Philippe?
A: The associated email addresses in account.pencil-code.org should be the same as what is registered in github as your primary email address.
2.7.7.6. Pencil Code discussion forum
Do I just need to send an email somewhere to subscribe or what?
A: The answer is yes; just go to:
http://groups.google.com/group/pencil-code-discuss
2.7.7.7. The manual
It would be a good idea to add this useful information in the manual, no?
A: When you have added new stuff to the code, don’t forget to mention
this in the pencil-code/doc/manual.tex file.
Again, the answer is yes; just go to:
cd pencil-code/doc/
vi manual.tex
svn ci -m "explanations about a new module in the code"
2.8. References
Abramowitz, A., & Stegun, I. A. (1984). Pocketbook of Mathematical Functions. Harri Deutsch, Frankfurt.
Babkovskaia, N., Haugen, N. E. L., & Brandenburg, A. (2011). J. Comp. Phys., 230(1), 12. A high-order public domain code for direct numerical simulations of turbulent combustion. (arXiv:1005.5301)
Barekat, A., & Brandenburg, A. (2014). Astron. Astrophys., 571, A68. “Near-polytropic stellar simulations with a radiative surface.”
Brandenburg, A. (2001). Astrophys. J., 550, 824–840. “The inverse cascade and nonlinear alpha-effect in simulations of isotropic helical hydromagnetic turbulence.”
Brandenburg, A. (2003). In Advances in non-linear dynamos (A. Ferriz-Mas & M. Núñez Jiménez, Eds.), The Fluid Mechanics of Astrophysics and Geophysics, Vol. 9, pp. 269–344. Taylor & Francis, London and New York. Available at http://arXiv.org/abs/astro-ph/0109497
Brandenburg, A., & Dobler, W. (2001). Astron. Astrophys., 369, 329–338. “Large scale dynamos with helicity loss through boundaries.”
Brandenburg, A., & Hazlehurst, J. (2001). Astron. Astrophys., 370, 1092–1102. “Evolution of highly buoyant thermals in a stratified layer.”
Brandenburg, A., & Kahniashvili, T. (2017). Phys. Rev. Lett., 118, 055102. “Classes of hydrodynamic and magnetohydrodynamic turbulent decay.”
Brandenburg, A., He, Y., Kahniashvili, T., Rheinhardt, M., & Schober, J. (2021). Astrophys. J., 911, 110. “Gravitational waves from the chiral magnetic effect.”
Brandenburg, A., & Sarson, G. R. (2002). Phys. Rev. Lett., 88, 055003. “The effect of hyperdiffusivity on turbulent dynamos with helicity.”
Brandenburg, A., Dobler, W., & Subramanian, K. (2002). Astron. Nachr., 323, 99–122. “Magnetic helicity in stellar dynamos: new numerical experiments.”
Brandenburg, A., Enqvist, K., & Olesen, P. (1996). Phys. Rev. D, 54, 1291–1300. “Large-scale magnetic fields from hydromagnetic turbulence in the very early universe.”
Brandenburg, A., Jennings, R. L., Nordlund, Å., Rieutord, M., Stein, R. F., & Tuominen, I. (1996). J. Fluid Mech., 306, 325–352. “Magnetic structures in a dynamo simulation.”
Brandenburg, A., Kahniashvili, T., Mandal, S., Roper Pol, A., Tevzadze, A. G., & Vachaspati, T. (2017). Phys. Rev. D, 96, 123528. “Evolution of hydromagnetic turbulence from the electroweak phase transition.”
Brandenburg, A., Moss, D., & Shukurov, A. (1995). MNRAS, 276, 651–662. “Galactic fountains as magnetic pumps.”
Brandenburg, A., Nordlund, Å., Stein, R. F., & Torkelsson, U. (1995). Astrophys. J., 446, 741–754. “Dynamo-generated turbulence and large scale magnetic fields in a Keplerian shear flow.”
Collatz, L. (1966). The numerical treatment of differential equations. Springer-Verlag, New York, p. 164.
Dobler, W., Stix, M., & Brandenburg, A. (2006). Astrophys. J., 638, 336–347. “Convection and magnetic field generation in fully convective spheres.”
Gammie, C. F. (2001). Astrophys. J., 553, 174–183. “Nonlinear outcome of gravitational instability in cooling, gaseous disks.”
Goodman, J., Narayan, R., & Goldreich, P. (1987). Month. Not. Roy. Soc., 225, 695–711. “The stability of accretion tori — II. Nonlinear evolution to discrete planets.”
Haugen, N. E. L., & Brandenburg, A. (2004). Phys. Rev. E, 70, 026405. “Inertial range scaling in numerical turbulence with hyperviscosity.”
Hockney, R. W., & Eastwood, J. W. (1981). Computer Simulation Using Particles. McGraw-Hill, New York.
Hurlburt, N. E., Toomre, J., & Massaguer, J. M. (1984). Astrophys. J., 282, 557–573. “Two-dimensional compressible convection extending over multiple scale heights.”
Kim, J., Moin, P., & Moser, R. (1987). J. Fluid Mech., 177, 133. “Turbulence statistics in fully developed channel flow at low Reynolds number.”
Kippenhahn, R., & Weigert, A. (1990). Stellar structure and evolution. Springer, Berlin.
Krause, F., & Rädler, K.-H. (1980). Mean-Field Magnetohydrodynamics and Dynamo Theory. Akademie-Verlag, Berlin; also Pergamon Press, Oxford.
Lele, S. K. (1992). J. Comp. Phys., 103, 16–42. “Compact finite difference schemes with spectral-like resolution.”
Misner, C. W., Thorne, K. S., & Wheeler, J. A. (1973). Gravitation. San Francisco: W. H. Freeman and Co., p. 213.
Mitra, D., Tavakol, R., Brandenburg, A., & Moss, D. (2009). Astrophys. J., 697, 923–933. “Turbulent dynamos in spherical shell segments of varying geometrical extent.” (arXiv:0812.3106)
Nordlund, Å., & Galsgaard, K. (1995). A 3D MHD code for Parallel Computers. Available at http://www.astro.ku.dk/~aake/NumericalAstro/papers/kg/mhd.ps.gz
Nordlund, Å., & Stein, R. F. (1990). Comput. Phys. Commun., 59, 119. “3-D simulations of solar and stellar convection and magnetoconvection.”
Olesen, P. (1997). Phys. Lett. B, 398, 321. “Inverse cascades and primordial magnetic fields.”
Press, W., Teukolsky, S., Vetterling, W., & Flannery, B. (1996). Numerical Recipes in Fortran 90 (2nd ed.). Cambridge.
Stanescu, D., & Habashi, W. G. (1988). J. Comp. Phys., 143, 674. “2N-storage low dissipation and dispersion Runge–Kutta schemes for computational acoustics.”
Williamson, J. H. (1980). J. Comp. Phys., 35, 48. “Low-storage Runge–Kutta schemes.”
Pencil Code Collaboration. (2021). J. Open Source Software, 6, 2807. “The Pencil Code, a modular MPI code for partial differential equations and particles: multipurpose and multiuser-maintained.”
Porter, T. A., Jóhannesson, G., & Moskalenko, I. V. (2022). Astrophys. J. Supp., 262, 30. “The GALPROP Cosmic-ray Propagation and Nonthermal Emissions Framework: Release v57.”
Intel. Fortran Compiler Developer Guide and Reference – mcmodel. https://software.intel.com/en-us/fortran-compiler-developer-guide-and-reference-mcmodel